HI-VQE Chemistry — funkcja Qiskit od Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Zobacz dokumentację API
Funkcje Qiskit to eksperymentalna funkcjonalność dostępna wyłącznie dla użytkowników planów IBM Quantum® Premium Plan, Flex Plan oraz On-Prem (za pośrednictwem IBM Quantum Platform API). Mają status wersji podglądowej i mogą ulec zmianie.
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
Omówienie
W chemii kwantowej problem struktury elektronowej polega na znalezieniu rozwiązań elektronowego równania Schrödingera — kwantowych funkcji falowych opisujących zachowanie elektronów w danym układzie. Funkcje te są wektorami amplitud zespolonych, przy czym każda amplituda odpowiada wkładowi jednej z możliwych konfiguracji elektronowych.
Stan podstawowy to funkcja falowa układu o najniższej energii i odgrywa szczególną rolę w badaniu układów molekularnych. Najbardziej dokładne podejście do obliczania stanu podstawowego uwzględnia wszystkie możliwe konfiguracje elektronowe, lecz dla większych układów staje się niewykonalne, ponieważ liczba konfiguracji rośnie wykładniczo wraz z rozmiarem układu.
Handover Iterative Variational Quantum Eigensolver (HI-VQE) to nowatorska hybrydowa metoda kwantowo-klasyczna służąca do precyzyjnego szacowania stanu podstawowego układów molekularnych. Łączy sprzęt kwantowy z obliczeniami klasycznymi — procesory kwantowe służą do efektywnego przeszukiwania kandydatów na konfiguracje elektronowe, a wynikowa funkcja falowa jest obliczana na komputerach klasycznych. Generując zwarte, lecz chemicznie dokładne funkcje falowe, HI-VQE wspomaga badania i odkrycia w chemii kwantowej oraz nauce o materiałach.

HI-VQE zmniejsza złożoność obliczeniową problemu struktury elektronowej, efektywnie szacując stan podstawowy z dużą dokładnością. Skupia się na starannie dobranym podzbiorze najważniejszych konfiguracji elektronowych, optymalizując jednocześnie dokładność i wydajność.
Łącząc zalety komputerów klasycznych i kwantowych, HI-VQE iteracyjnie udoskonala bieżące przybliżenie funkcji falowej. Unikalne techniki budowy podprzestrzeni pomagają zwiększyć efektywność doboru konfiguracji, zapewniając użytkownikom większą kontrolę obliczeniową i lepszą dokładność symulacji chemii kwantowej.
Jeśli chcesz dokładniej poznać algorytm, możesz przeczytać powiązaną publikację naukową.
Opis
Liczba konfiguracji elektronowych w układzie molekularnym rośnie wykładniczo wraz z rozmiarem układu. Jednak dla pewnych stanów elektronowych, takich jak stan podstawowy, zazwyczaj tylko niewielki ułamek konfiguracji wnosi istotny wkład do energii stanu. Metody wybranej interakcji konfiguracyjnej (SCI) wykorzystują tę rzadkość do obniżenia kosztów obliczeniowych, identyfikując i koncentrując się na najbardziej istotnych konfiguracjach. Podzbiór tych konfiguracji określa się mianem podprzestrzeni.
HI-VQE wykorzystuje wrodzoną efektywność komputerów kwantowych w reprezentowaniu układów molekularnych, wspomagając przeszukiwanie podprzestrzeni. Integruje klasyczne i kwantowe procedury, aby rozwiązać problem struktury elektronowej z dużą dokładnością. W odróżnieniu od istniejących kwantowych metod SCI, HI-VQE łączy trenowanie wariacyjne, iteracyjne budowanie podprzestrzeni oraz wstępne przesiewanie konfiguracji metodą pre-diagonalizacji, co zwiększa efektywność przez ograniczenie liczby pomiarów kwantowych, iteracji i kosztów klasycznej diagonalizacji. HI-VQE można zatem stosować do większych układów molekularnych wymagających większej liczby Qubitów, a koszt rozwiązania problemu danego rozmiaru z tym samym stopniem dokładności ulega zmniejszeniu.
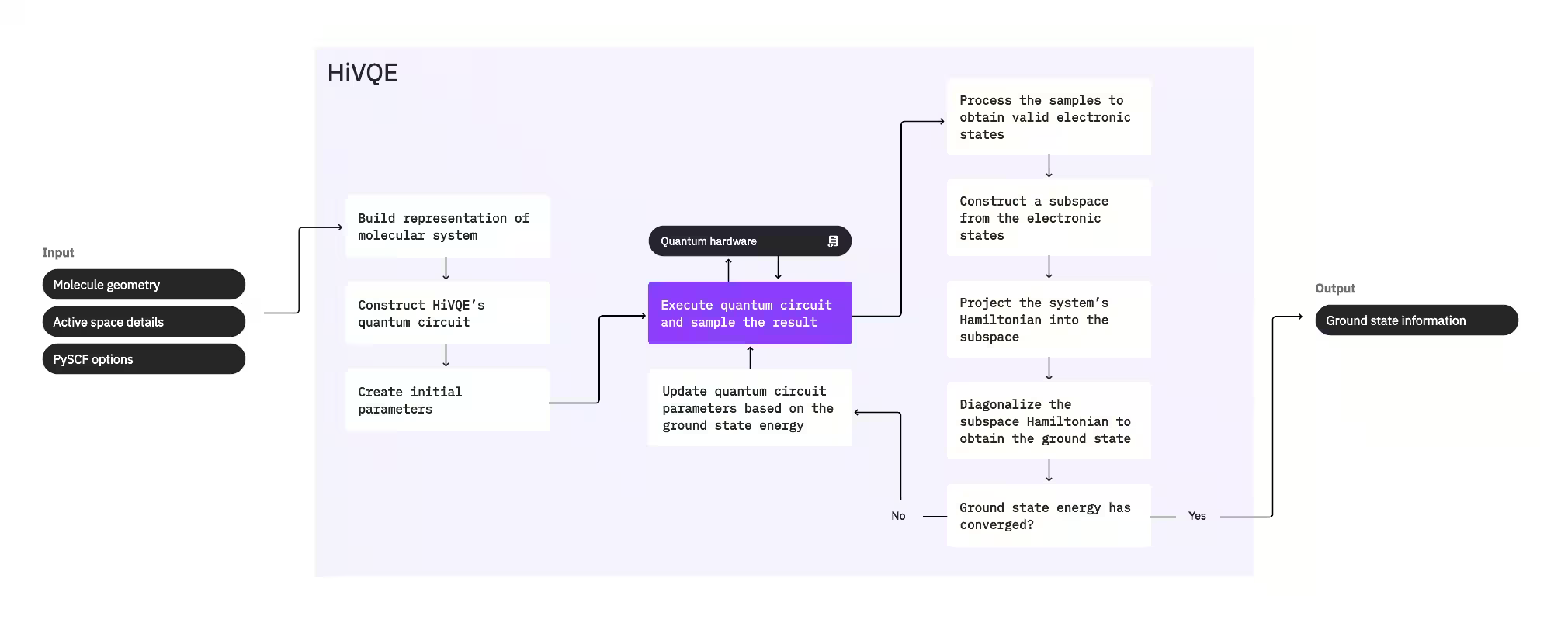
Aby obliczyć stan podstawowy układu, HI-VQE najpierw używa klasycznego pakietu chemicznego PySCF do wygenerowania reprezentacji molekularnej na podstawie danych wejściowych podanych przez użytkownika, takich jak geometria molekularna i inne informacje o cząsteczce. Następnie wchodzi w hybrydową pętlę optymalizacji kwantowo-klasycznej, iteracyjnie udoskonalając podprzestrzeń, by optymalnie reprezentować stan podstawowy przy minimalizacji liczby zawartych konfiguracji. Pętla działa do momentu spełnienia kryteriów zbieżności, takich jak rozmiar podprzestrzeni lub stabilność energii; po ich osiągnięciu wynikiem jest obliczona funkcja falowa stanu podstawowego wraz z energią. Wyniki te można wykorzystać do konstruowania dokładnych powierzchni energii potencjalnej i przeprowadzania dalszej analizy układu.
Pętla optymalizacji skupia się na dostosowywaniu parametrów Circuit kwantowego w celu wygenerowania wysokiej jakości podprzestrzeni. HI-VQE oferuje trzy opcje Circuit kwantowego: excitation_preserving, efficient_su2 oraz LUCJ. Optymalizacja jest inicjalizowana blisko stanu referencyjnego Hartree-Focka ze względu na jego ogólną przydatność. Następnie Circuit jest wykonywany na urządzeniu kwantowym i z wynikowego stanu kwantowego pobierane są próbki konfiguracji, które są zwracane jako ciągi binarne. Ze względu na szumy urządzenia kwantowego niektóre próbkowane konfiguracje mogą być fizycznie nieprawidłowe — nie zachowują liczby elektronów ani spinu. HI-VQE rozwiązuje ten problem, stosując proces odtwarzania konfiguracji z pakietu qiskit-addon-sqd, dzięki czemu użytkownicy mogą albo poprawiać nieprawidłowe konfiguracje, albo je odrzucać.
Prawidłowe konfiguracje przechodzą opcjonalny krok przesiewania, usuwający te, które przewiduje się jako wnoszące minimalny wkład. Zmniejsza to wymiar podprzestrzeni, obniżając tym samym koszt kroku diagonalizacji. Jeśli przesiewanie jest włączone, na podstawie prawidłowych konfiguracji konstruowany jest wstępny hamiltonian podprzestrzeni, a diagonalizacja wykonywana jest z bardzo luźnymi kryteriami zakończenia. Choć dokładność wynikowych amplitud dla każdej konfiguracji jest niska, jest ona skuteczna do przewidywania, które konfiguracje pominąć w podprzestrzeni w danej iteracji, i jest szybka do obliczenia.
Wybrane konfiguracje są dodawane do podprzestrzeni, a hamiltonian układu jest rzutowany do tej podprzestrzeni. Podprzestrzeń aktualizuje się iteracyjnie, zachowując najważniejsze konfiguracje pomiędzy iteracjami. Podejście to różni się od metod alternatywnych tym, że Circuit kwantowy nie musi przybliżać pełnego stanu podstawowego w każdym kroku.
Następnie hamiltonian podprzestrzeni jest klasycznie diagonalizowany w celu uzyskania najniższej wartości własnej i odpowiadającego jej wektora własnego, reprezentującego przybliżenie stanu podstawowego i jego energii. W miarę jak jakość podprzestrzeni poprawia się w kolejnych iteracjach, obliczony stan podstawowy coraz lepiej przybliża rzeczywisty stan podstawowy. Na tym etapie można przeprowadzić dodatkowy krok przesiewania, usuwający z podprzestrzeni wszelkie konfiguracje, które nie wnoszą istotnego wkładu do obliczonego stanu podstawowego. Krok ten zapewnia, że podprzestrzeń przenoszona do następnej iteracji jest możliwie jak najbardziej zwarta. Ocena odbywa się na podstawie amplitud zwróconych przez diagonalizację, gdyż reprezentują one ważność wkładu każdej konfiguracji do obliczonego stanu podstawowego.
Sprawdzenie zbieżności rozstrzyga, czy dalsze trenowanie mogłoby poprawić wyniki. Jeśli tak, wykonywany jest opcjonalny krok klasycznej ekspansji, parametry Circuit kwantowego są aktualizowane w celu dalszej minimalizacji obliczonej energii, a proces się powtarza. Krok klasycznej ekspansji generuje dodatkowe konfiguracje dla podprzestrzeni, uzupełniając konfiguracje próbkowane z urządzenia kwantowego. Najpierw identyfikuje konfigurację o największej amplitudzie w wynikach diagonalizacji, a następnie generuje nowe konfiguracje z pojedynczymi i podwójnymi wzbudzeniami z tej konfiguracji. Żądana liczba tych konfiguracji jest następnie dodawana do podprzestrzeni.
Gdy zostanie stwierdzona zbieżność iteracji, HI-VQE zwraca obliczony stan podstawowy (w postaci stanów w podprzestrzeni i ich amplitud w funkcji falowej stanu podstawowego), jego energię oraz miarę wariancji energii, która wskazuje, czy obliczony stan stanowi stan własny hamiltonianu układu.
Użytkownicy mogą decydować o używanym Circuit kwantowym i liczbie strzałów (shots) dla każdego Circuit kwantowego, a także kontrolować rozmiar podprzestrzeni lub włączyć klasyczne generowanie dodatkowych konfiguracji wspomagających konfiguracje generowane kwantowo. Dzięki temu użytkownicy mogą dostosować zachowanie HI-VQE do swoich docelowych zastosowań.
Licencjonowanie
Pamiętaj, że korzystanie z tej funkcji Qiskit jest ograniczone do problemów wymagających co najwyżej 20 Qubitów, chyba że uzyskano licencję przyznającą wyższy limit.
Jeśli chcesz uzyskać informacje na temat licencji, napisz na adres qiskit.support@qunovacomputing.com.
Pierwsze kroki
Najpierw poproś o dostęp do funkcji. Następnie uwierzytelnij się przy użyciu swojego klucza API IBM Quantum® i — zakładając, że masz już zapisane konto w środowisku lokalnym — załaduj funkcję Qiskit w następujący sposób:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Przykład
Pierwszy przykład pokazuje, jak obliczyć energię stanu podstawowego cząsteczki NH3 przy użyciu algorytmu HI-VQE.
Zdefiniuj geometrię molekularną i opcje
Geometria molekularna NH3 jest podana we współrzędnych kartezjańskich oddzielonych znakiem ";" dla każdego atomu.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Dodatkowe opcje można zdefiniować i przekazać dla układu molekularnego w następującym formacie słownika.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Uruchom funkcję, podając geometrię i opcje jako dane wejściowe.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Dobrym pomysłem jest wydrukowanie identyfikatora zadania Function, aby móc go podać w zgłoszeniu do pomocy technicznej, jeśli coś pójdzie nie tak.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Ten przykład wykorzystuje 16 Qubitów z 8 orbitalami bazy sto3g dla cząsteczki NH3. Sprawdź status zadania Qiskit Function lub pobierz wyniki w następujący sposób:
print(job.status())
QUEUED
Po zakończeniu zadania wyniki można uzyskać za pomocą instancji result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Aby uzyskać dostęp do energii stanu podstawowego, użyj klucza "energy". Klucz "eigenvector" dostarcza współczynniki CI wraz z odpowiadającą im notacją bitstring konfiguracji elektronowej, przechowywaną pod kluczem "states" wyników.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Wydajność
W tej sekcji przedstawiono wyniki wzorcowych obliczeń HI-VQE: przypadek 24-Qubitowy dla Li2S, przypadek 40-Qubitowy dla cząsteczki N2 oraz przypadek 44-Qubitowy dla układu FeP-NO.
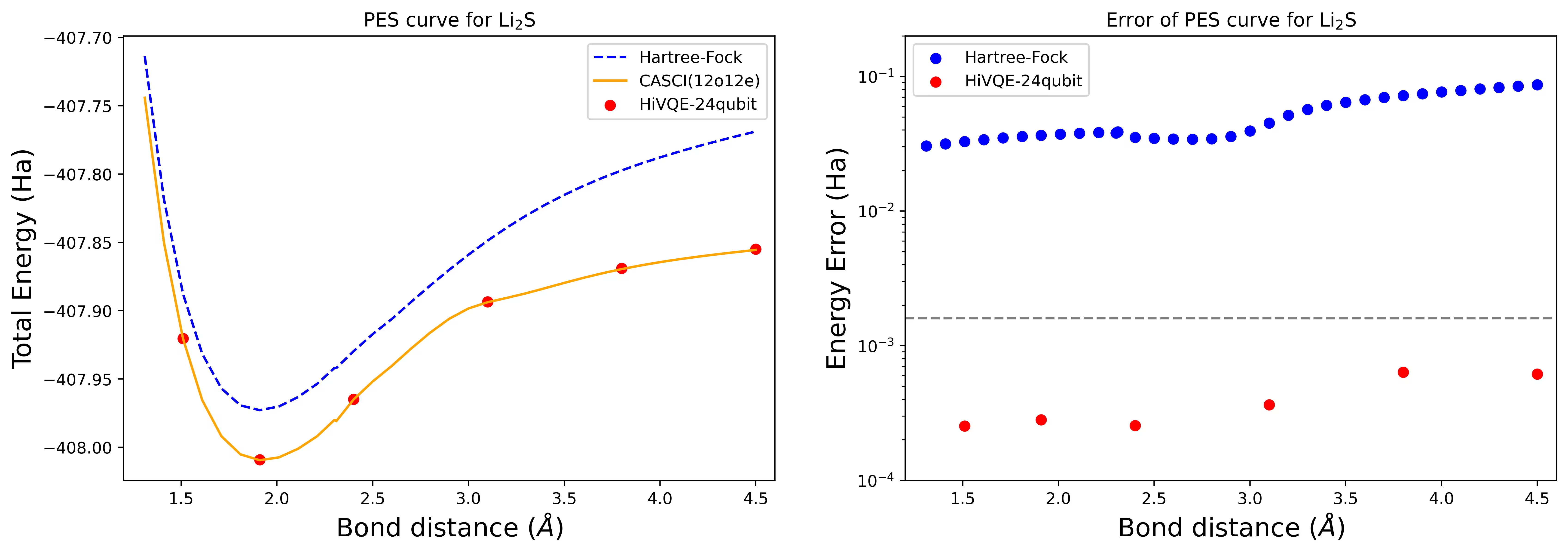
Krzywa potencjalnej powierzchni energii dysocjacji dla cząsteczki Li2S z 24 Qubitami
Krzywa PES jest pokazana wraz z referencją FCI i początkowym przybliżeniem z RHF, a także błędem energii względem referencji FCI.
 .
.
Obliczenia zostały przeprowadzone dla następujących geometrii i opcji.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Czerwone punkty reprezentują wyniki obliczeń HI-VQE dla sześciu różnych geometrii; trzy geometrie odpowiadające odległościom 1,51, 2,40 i 3,80 angstrema podano jako dane wejściowe w powyższej komórce.
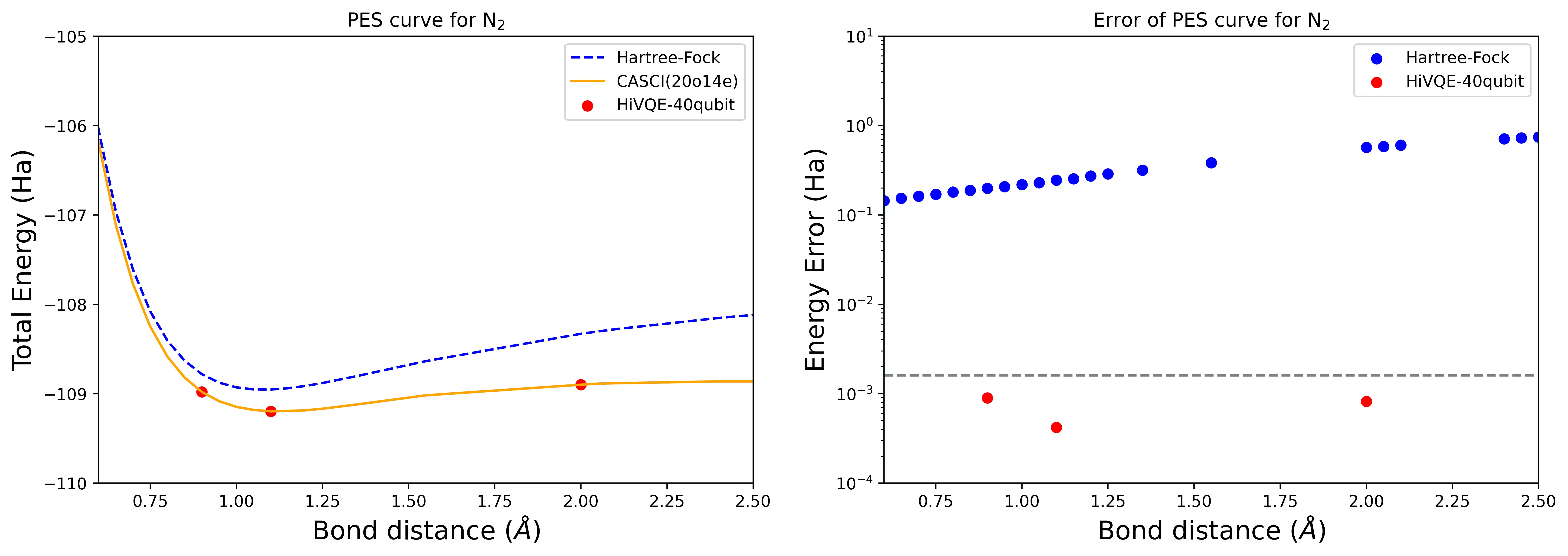
Krzywa PES dysocjacji dla cząsteczki N2 z 40 Qubitami
Cząsteczka azotu została zidentyfikowana jako układ wieloreferencyjny z dużymi wkładami energii korelacyjnej wykraczającymi poza stan Hartree-Focka. Przeprowadzono obliczenia wzorcowe dla cząsteczki N2 z bazą cc-pvdz, (20o,14e) z wykorzystaniem homo-lumo do wyboru aktywnych orbitali. Liczba stanów reprezentujących ten problem w pełnej przestrzeni aktywnej (CAS) wynosi 6 009 350 400. Rozwiązanie zagadnienia własnego (dla energii i struktury elektronowej) przy tej liczbie stanów nie jest możliwe nawet na wydajnym komputerze stacjonarnym (16 CPU/64 GB). Dzięki HI-VQE możesz efektywnie przeszukiwać podprzestrzeń stanów CAS, uzyskując wyniki o chemicznej dokładności przy znacznie mniejszym zużyciu zasobów obliczeniowych. Poniższe wykresy przedstawiają krzywą PES obliczoną metodą HI-VQE (40 Qubitów) dla dysocjacji cząsteczki N2.
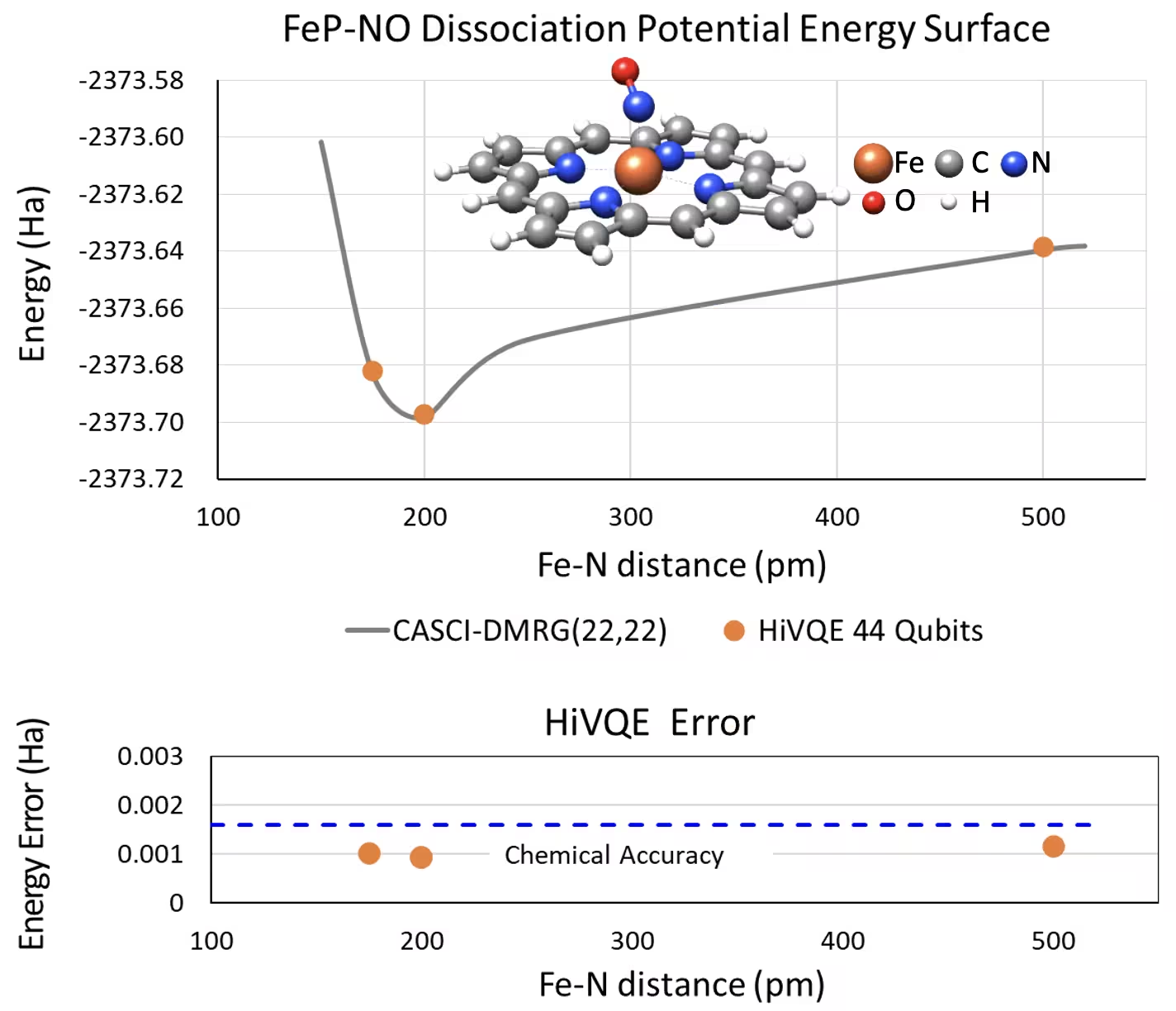
Krzywa PES dysocjacji dla pięciokoordynacyjnego żelaza(II)-porfiryny z układem NO z 44 Qubitami
Innym interesującym układem chemicznym jest kompleks żelaza(II)-porfiryny (FeP) z skoordynowanym tlenkiem azotu (NO) jako ligandem — jest to biologicznie istotny układ metaloporfiryny odgrywający kluczową rolę w różnych procesach fizjologicznych. W tym przykładzie HI-VQE zostało wykorzystane do oszacowania dokładnej krzywej potencjalnej powierzchni energii oddziaływania międzycząsteczkowego między FeP a NO (energia stanu podstawowego dla geometrii o różnych odległościach). Połączony układ ma 450 orbitali i 202 elektrony (450o,202e) z bazą 6-31g(d). Do obliczenia mniejszego przypadku z rzeczywistego układu (22o,22e) zastosowano selekcję aktywnych orbitali homo-lumo. Na podstawie poniższych wyników wzorcowych udało się osiągnąć dokładność chemiczną (> 1,6 mHa) w odniesieniu do klasycznych obliczeń CASCI(DMRG) (22o,22e).
Testy wydajnościowe
- Dokładny rozmiar macierzy to liczba determinantów dla dokładnego rozwiązania, takiego jak FCI i CASCI.
- HI-VQE próbkuje i oblicza podprzestrzeń tej macierzy (tj. rozmiar macierzy HI-VQE).
- Całkowity czas obejmuje czas pracy QPU oraz uruchomienia Qiskit Function z procesorem CPU.
- Dokładność jest szacowana na podstawie różnicy energii od dokładnego rozwiązania.
| Układ chemiczny | Liczba Qubitów | Dokładny rozmiar macierzy | Rozmiar macierzy HI-VQE | E(różn.) od dokładnego (mHa) | Liczba iteracji | Całkowity czas | Czas pracy QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0,08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0,60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0,90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1,10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0,90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1,20 | 21 | 46547 s | 258 s |
Pobieranie komunikatów o błędach
Jeśli twoje zadanie zakończy się niepowodzeniem, status będzie miał wartość ERROR, a wywołanie job.result() spowoduje zgłoszenie wyjątku:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Uzyskaj wsparcie
Możesz wysłać wiadomość e-mail na adres qiskit.support@qunovacomputing.com, aby uzyskać pomoc dotyczącą tej funkcji.
Jeśli potrzebujesz pomocy w rozwiązaniu konkretnego błędu, podaj identyfikator zadania Function, w którym wystąpił błąd.
Następne kroki
- Poproś o dostęp do funkcji, wypełniając ten formularz.
- Odwiedź dokumentację API tej funkcji Qiskit.
- Wypróbuj samouczek Obliczanie krzywej PES dysocjacji dla FeP-NO z HI-VQE.
- Zapoznaj się z Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Wypróbuj samouczek Krzywe PES dysocjacji z Qunova HiVQE.