Diagonalizacja kwantowa oparta na próbkowaniu hamiltonianów chemicznych
Szacowany czas użycia: poniżej jednej minuty na procesorze Heron r2 (UWAGA: jest to wyłącznie szacunek. Rzeczywisty czas wykonania może się różnić.)
Cele kształcenia
Po ukończeniu tego samouczka użytkownicy powinni rozumieć:
- Jak używać dodatku SQD do Qiskit do aproksymacji energii stanu podstawowego układu molekularnego za pomocą bitciągów próbkowanych z jednostki przetwarzania kwantowego (QPU).
- Jak używać ffsim do konstruowania obwodu ansatz lokalnego unitarnego klastera Jastrow (LUCJ) do symulacji chemii kwantowej.
Wymagania wstępne
Zalecamy, aby użytkownicy zapoznali się z następującymi tematami przed przystąpieniem do tego samouczka:
- Chemia kwantowa i druga kwantyzacja
- Używanie prymitywu Sampler do próbkowania z obwodów kwantowych
Tło
W tym samouczku pokazujemy, jak przetwarzać zaszumione próbki kwantowe w celu aproksymacji stanu podstawowego cząsteczki azotu przy równowagowej długości wiązania, używając dodatku SQD do Qiskit do implementacji algorytmu diagonalizacji kwantowej opartej na próbkowaniu (SQD). Więcej szczegółów dotyczących oprogramowania można znaleźć w odpowiedniej dokumentacji, w tym prosty przykład na początek.
Ten samouczek jest zalecany dla użytkowników zaznajomionych z chemią kwantową: konkretnie, dla osób znających metody znajdowania energii stanu podstawowego cząsteczki. Szczegółowy opis przepływu pracy znajdziesz w kursie algorytmu diagonalizacji kwantowej.
SQD to technika znajdowania wartości własnych i wektorów własnych operatorów kwantowych, takich jak hamiltonian układu kwantowego, łącząca obliczenia kwantowe i rozproszone klasyczne. Rozproszone obliczenia klasyczne służą do przetwarzania próbek uzyskanych z procesora kwantowego oraz do rzutowania i diagonalizacji docelowego hamiltonianu w podprzestrzeni przez nie rozpinanej. Przepływ pracy oparty na SQD składa się z następujących kroków:
- Wybierz ansatz obwodu i zastosuj go na komputerze kwantowym do stanu odniesienia (w tym przypadku stanu Hartree-Focka).
- Próbkuj bitciągi z wynikowego stanu kwantowego.
- Uruchom procedurę samospójnego odtwarzania konfiguracji na bitciągach, aby uzyskać aproksymację stanu podstawowego.
Wiadomo, że SQD działa dobrze, gdy docelowy stan własny jest rzadki: funkcja falowa jest wspierana przez zbiór stanów bazowych , którego rozmiar nie rośnie wykładniczo wraz z rozmiarem problemu.
Chemia kwantowa
Hamiltonian układu molekularnego można zapisać jako
gdzie i są liczbami zespolonymi zwanymi całkami molekularnymi, które można obliczyć na podstawie specyfikacji cząsteczki za pomocą programu komputerowego. W tym samouczku obliczamy całki za pomocą pakietu oprogramowania PySCF.
Szczegóły dotyczące wyprowadzenia hamiltonianu molekularnego znajdziesz w podręczniku chemii kwantowej (na przykład Modern Quantum Chemistry autorstwa Szabo i Ostlunda). Wysokopoziomowe wyjaśnienie, jak problemy chemii kwantowej są odwzorowywane na komputery kwantowe, znajdziesz w wykładzie Mapping Problems to Qubits z Qiskit Global Summer School 2024.
Ansatz lokalnego unitarnego klastera Jastrow (LUCJ)
SQD wymaga ansatzu obwodu kwantowego do pobierania próbek. W tym samouczku użyjemy ansatzu lokalnego unitarnego klastera Jastrow (LUCJ) ze względu na połączenie motywacji fizycznej i przyjazności sprzętowej. Do skonstruowania obwodu ansatzu użyjemy ffsim.
Ansatz LUCJ dostosowuje się do QPU z ograniczoną łącznością kubitów. Orbitale spinowe są odwzorowywane na kubity tak, aby ansatz nie wymagał routingu bramkami SWAP. Sprzęt IBM® ma topologię kubitów z siatką heavy-hex, w której możemy przyjąć wzorzec „zygzak", przedstawiony poniżej. W tym wzorcu orbitale o tym samym spinie są odwzorowane na kubity o topologii liniowej (czerwone i niebieskie kółka), a połączenie między orbitalami różnych spinów występuje co 4. orbital przestrzenny, przy czym połączenie jest realizowane za pomocą kubitu pomocniczego (fioletowe kółka).
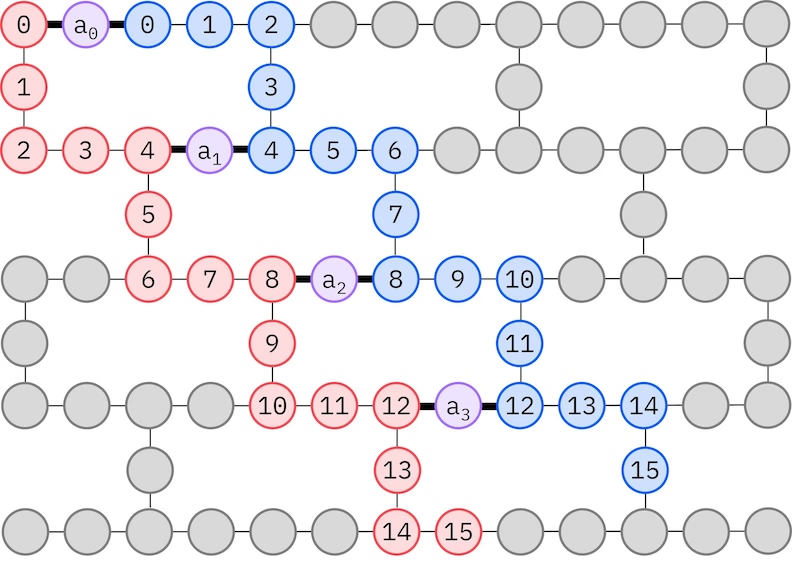
Samospójne odtwarzanie konfiguracji
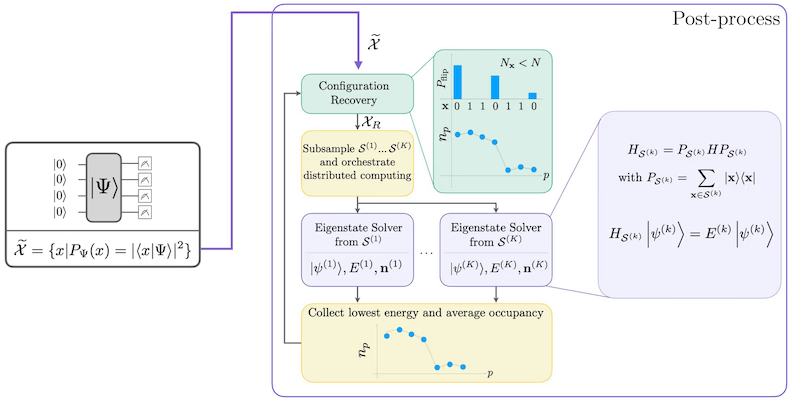
Procedura samospójnego odtwarzania konfiguracji jest zaprojektowana tak, aby wydobyć jak najwięcej sygnału z zaszumionych próbek kwantowych. Ponieważ hamiltonian molekularny zachowuje liczbę cząstek i spin Z, sensowne jest wybranie ansatzu obwodu, który również zachowuje te symetrie. Gdy zostanie zastosowany do stanu Hartree-Focka, wynikowy stan ma stałą liczbę cząstek i spin Z w bezszumowym przypadku. Dlatego spino- i spino- połowy dowolnego bitciągu próbkowanego z tego stanu powinny mieć tę samą wagę Hamminga co w stanie Hartree-Focka. Z powodu obecności szumu w obecnych procesorach kwantowych niektóre zmierzone bitciągi będą naruszać tę właściwość. Prostą formą postselekcji byłoby odrzucenie tych bitciągów, ale jest to marnotrawstwo, ponieważ te bitciągi mogą nadal zawierać pewien sygnał. Procedura samospójnego odtwarzania próbuje odzyskać część tego sygnału w przetwarzaniu końcowym. Procedura jest iteracyjna i wymaga jako danych wejściowych oszacowania średnich obsadzeń każdego orbitalu w stanie podstawowym, które są najpierw obliczane na podstawie surowych próbek. Procedura jest uruchamiana w pętli, a każda iteracja składa się z następujących kroków:
- Dla każdego bitciągu naruszającego określone symetrie odwróć jego bity za pomocą procedury probabilistycznej zaprojektowanej tak, aby przybliżyć bitciąg do bieżącego oszacowania średnich obsadzeń orbitalnych, uzyskując nowy bitciąg.
- Zbierz wszystkie stare i nowe bitciągi spełniające symetrie i pobierz podzestawy o ustalonym z góry rozmiarze.
- Dla każdego podzbioru bitciągów rzutuj hamiltonian na podprzestrzeń rozpinaną przez odpowiednie wektory bazowe (opis tych wektorów bazowych znajdziesz w poprzedniej sekcji) i oblicz aproksymację stanu podstawowego rzutowanego hamiltonianu na komputerze klasycznym.
- Zaktualizuj oszacowanie średnich obsadzeń orbitalnych za pomocą aproksymacji stanu podstawowego o najniższej energii.
Diagram przepływu pracy SQD
Przepływ pracy SQD jest przedstawiony na poniższym diagramie:
Wymagania
Przed rozpoczęciem tego samouczka upewnij się, że masz zainstalowane:
- Qiskit SDK v1.0 lub nowszy, z obsługą wizualizacji
- Qiskit Runtime v0.22 lub nowszy (
pip install qiskit-ibm-runtime) - Dodatek SQD do Qiskit v0.11 lub nowszy (
pip install qiskit-addon-sqd) - ffsim v0.0.75 lub nowszy (
pip install ffsim)
Konfiguracja
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Przykład na małą skalę z symulatorem
W tym samouczku znajdziemy aproksymację stanu podstawowego cząsteczki azotu w pobliżu równowagowej odległości wiązania. Najpierw używamy małego zestawu bazowego STO-6G, abyśmy mogli zasymulować eksperyment i sprawdzić, czy działa poprawnie.
Krok 1: Odwzorowanie klasycznych danych wejściowych na problem kwantowy
Najpierw określamy właściwości cząsteczki i jej parametry.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Przed skonstruowaniem obwodu ansatzu LUCJ najpierw wykonujemy obliczenie CCSD w poniższej komórce kodu. Amplitudy i z tego obliczenia zostaną użyte do inicjalizacji parametrów ansatzu.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Teraz używamy ffsim do tworzenia obwodu ansatzu. Ponieważ nasza cząsteczka ma zamknięto-powłokowy stan Hartree-Focka, używamy zrównoważonego spinowo wariantu ansatzu UCJ, UCJOpSpinBalanced. Ustawiamy optimize=True w metodzie from_t_amplitudes, aby włączyć „skompresowaną" podwójną faktoryzację amplitud (szczegóły znajdziesz w The local unitary cluster Jastrow (LUCJ) ansatz w dokumentacji ffsim).
Ponieważ ansatz LUCJ dostosowuje się do dostępnej łączności QPU, musimy zainicjować backend QPU przed jego utworzeniem. Na razie stworzymy ogólny backend z mapą sprzężeń heavy-hex i zestawem bramek, do którego ansatz LUCJ naturalnie się rozkłada. Następnie użyjemy ffsim.qiskit.generate_lucj_pass_manager, aby stworzyć menedżer przejść wyspecjalizowany do transpilacji ansatzu LUCJ na dany backend zgodnie z układem „zygzak" opisanym w sekcji tła dotyczącej ansatzu LUCJ. Ta funkcja używa heurystyki oceniającej, aby zminimalizować błędy związane z wybranym układem, co jest ważne, jeśli twój backend to prawdziwy QPU lub symulator z modelem szumu. Oprócz zwracania menedżera przejść, ta funkcja zwraca również pary sprzężeń alfa-beta, które można zrealizować na sprzęcie. Jeśli nie wszystkie pary mogą zostać zrealizowane, emituje ostrzeżenie.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Krok 2: Optymalizacja pod kątem wykonania na sprzęcie kwantowym
Następnie optymalizujemy obwód dla docelowego sprzętu. Zazwyczaj ten krok obejmuje inicjalizację backendu sprzętowego i menedżera przejść dla tego backendu. Jednak ponieważ ansatz LUCJ jest dostosowany do łączności sprzętu, już wykonaliśmy te czynności w poprzednim kroku. Pozostało tylko uruchomić menedżer przejść na obwodzie, aby transpilować go do obwodu ISA, który może być bezpośrednio wykonany na QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Krok 3: Wykonanie za pomocą prymitywów Qiskit
Po zoptymalizowaniu obwodu pod kątem wykonania na sprzęcie jesteśmy gotowi uruchomić go na docelowym sprzęcie i zebrać próbki do estymacji energii stanu podstawowego. Ponieważ mamy tylko jeden obwód, użyjemy trybu wykonania zadań Qiskit Runtime i wykonamy nasz obwód.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Krok 4: Przetwarzanie końcowe i zwrócenie wyniku w żądanym formacie klasycznym
Przydatną miarą oceny jakości danych wyjściowych QPU jest liczba zwróconych prawidłowych konfiguracji. Prawidłowa konfiguracja ma właściwą liczbę cząstek i spin Z, co oznacza, że prawa połowa bitciągu ma wagę Hamminga równą liczbie elektronów ze spinem w górę, a lewa połowa ma wagę Hamminga równą liczbie elektronów ze spinem w dół. Poniższa komórka oblicza frakcję spróbkowanych konfiguracji, które są prawidłowe.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Wszystkie bitciągi są prawidłowe, ponieważ próbkujemy obwód na bezszumowym symulatorze. Podczas działania na zaszumionym QPU frakcja będzie mniejsza niż jeden, ale miejmy nadzieję, że będzie większa niż frakcja, której można by oczekiwać, gdyby bitciągi były próbkowane równomiernie losowo, co jest obliczane w poniższej komórce.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Teraz estymujemy energię stanu podstawowego hamiltonianu za pomocą funkcji diagonalize_fermionic_hamiltonian. Funkcja ta wykonuje procedurę samospójnego odtwarzania konfiguracji, aby iteracyjnie udoskonalać zaszumione próbki kwantowe w celu poprawy estymacji energii. Przekazujemy funkcję zwrotną, abyśmy mogli zapisać wyniki pośrednie do późniejszej analizy. Wyjaśnienia argumentów funkcji diagonalize_fermionic_hamiltonian znajdziesz w dokumentacji API.
Tutaj używamy argumentu initial_occupancies funkcji diagonalize_fermionic_hamiltonian, aby określić konfigurację Hartree-Focka jako wstępne oszacowanie obsadzeń orbitalnych w stanie podstawowym. Takie podejście jest sensowne dla układów, w których stan podstawowy ma znaczące wsparcie na konfiguracji Hartree-Focka, ale może nie być odpowiednie w innych sytuacjach, choć bardziej zaawansowane metody obliczeniowe mogą w takich przypadkach dawać lepsze wstępne oszacowania. Określenie initial_occupancies pozwala również na uruchomienie odtwarzania konfiguracji nawet wtedy, gdy nie spróbkowano żadnych prawidłowych konfiguracji, co może mieć miejsce podczas próbkowania dużego obwodu na zaszumionym QPU. Bez tego argumentu odtwarzanie konfiguracji zakończyłoby się niepowodzeniem i zwróciłoby błąd, jeśli nie dostarczono żadnych prawidłowych konfiguracji.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
Wizualizacja wyników
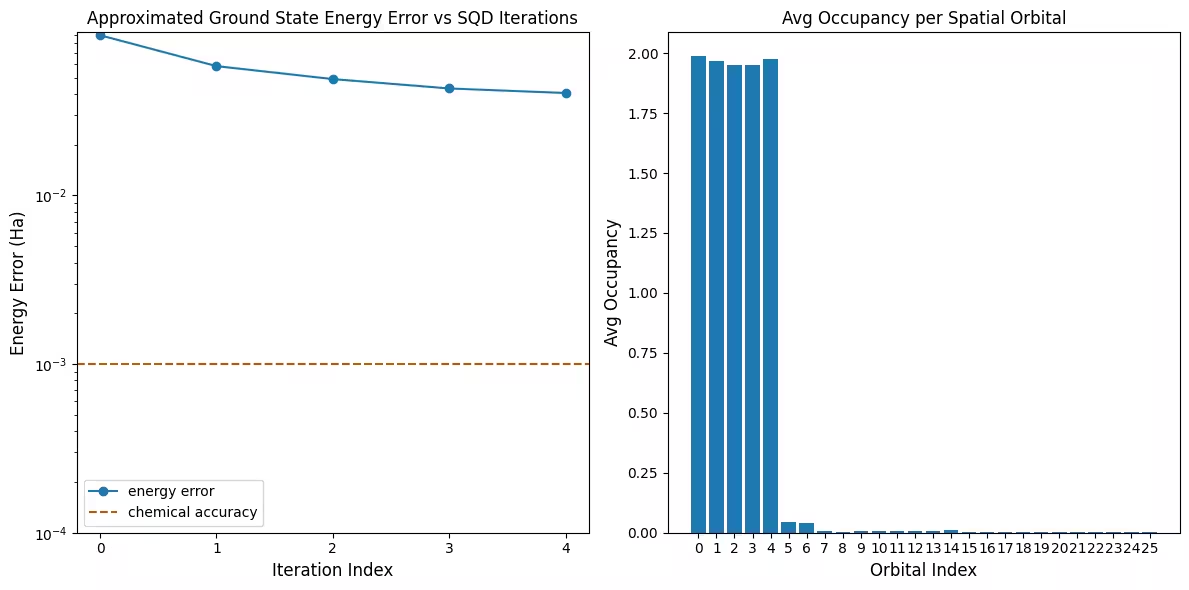
Pierwszy wykres pokazuje, że w tej symulacji jesteśmy już w granicach 1 mH od dokładnej odpowiedzi po pierwszej iteracji (dokładność chemiczna jest zazwyczaj przyjmowana jako 1 kcal/mol 1.6 mH). To jest mały układ, a ponieważ próbki są bezszumowe, odtwarzanie konfiguracji nie jest potrzebne. W przypadku większego układu działającego na zaszumionym QPU może być potrzebnych wiele iteracji odtwarzania konfiguracji, a końcowa dokładność może być gorsza. Ogólnie energię można poprawić, dopuszczając więcej iteracji odtwarzania konfiguracji lub zwiększając liczbę próbek na partię.
Drugi wykres pokazuje średnie obsadzenie każdego orbitalu przestrzennego po ostatniej iteracji. Widzimy, że zarówno elektrony ze spinem w górę, jak i ze spinem w dół zajmują pięć pierwszych orbitali z dużym prawdopodobieństwem w naszych rozwiązaniach.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Przykład na dużą skalę na rzeczywistym sprzęcie
Teraz uruchamiamy większy przykład na rzeczywistym sprzęcie kwantowym. Tutaj wyprowadzamy przestrzeń aktywną dla cząsteczki azotu z zestawu bazowego cc-pVDZ.
Kroki 1-4
Tutaj łączymy wszystkie kroki w jeden przepływ pracy na większą skalę, który jest następnie uruchamiany na rzeczywistym sprzęcie kwantowym.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Następne kroki
Jeśli ta praca wydała ci się interesująca, możesz zainteresować się następującymi materiałami:
- Kwantowa diagonalizacja Krylova oparta na próbkowaniu modelu sieciowego fermionów - powiązany samouczek wykorzystujący obwody ewolucji czasowej zamiast ansatzu wariacyjnego
- Skalowanie przepływów pracy chemii SQD z rozwiązaniem Dice - strona pokazująca, jak używać bardziej wydajnego oprogramowania Dice do diagonalizacji
- Dokumentacja API dodatku SQD - dokumentacja funkcji
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - artykuł, na którym oparty jest ten samouczek