Diagonalizacja kwantowa Krylova oparta na próbkowaniu dla fermionowego modelu sieciowego
Szacowane użycie: Dziewięć sekund na procesorze Heron r2 (UWAGA: To jest tylko szacunek. Twój czas wykonania może być inny.)
Efekty uczenia się
Po przejściu przez ten samouczek użytkownicy powinni rozumieć:
- Jak używać dodatku SQD do Qiskit, aby przybliżyć energię stanu podstawowego modelu sieciowego przy użyciu bitstrings próbkowanych z jednostki przetwarzania kwantowego (QPU).
- Jak używać ffsim do konstruowania obwodów ewolucji czasowej dla symulacji fermionowej.
- Jak łączyć próbki z wielu obwodów do post-processingu przy użyciu algorytmu diagonalizacji kwantowej Krylova opartej na próbkowaniu (SKQD).
Wymagania wstępne
Sugerujemy, aby użytkownicy zaznajomili się z następującymi tematami przed przystąpieniem do tego samouczka:
- Kwantowa diagonalizacja oparta na próbkowaniu hamiltonowskiego chemicznego
- Diagonalizacja kwantowa Krylova dla hamiltonianów sieciowych
- Prymitywy Qiskit
Tło
Ten samouczek pokazuje, jak używać kwantowej diagonalizacji opartej na próbkowaniu (SQD) do szacowania energii stanu podstawowego fermionowego modelu sieciowego. Konkretnie badamy jednowymiarowy jednodomieszkowy model Andersona (SIAM), który służy do opisu magnetycznych domieszek osadzonych w metalach.
Ten samouczek podąża za podobnym przepływem pracy co powiązany samouczek Kwantowa diagonalizacja oparta na próbkowaniu hamiltonowskiego chemicznego. Jednak kluczowa różnica leży w sposobie budowania obwodów kwantowych. Drugi samouczek używa heurystycznego wariacyjnego ansatzu, który jest atrakcyjny dla hamiltonianów chemicznych potencjalnie z milionami wyrazów oddziaływań. Z drugiej strony, ten samouczek używa obwodów przybliżających ewolucję czasową przez hamiltonian. Takie obwody mogą być głębokie, co sprawia, że to podejście jest lepsze dla zastosowań do modeli sieciowych. Wektory stanów przygotowane przez te obwody tworzą bazę dla podprzestrzeni Krylova, a w rezultacie algorytm udowodnialnie i efektywnie zbiega do stanu podstawowego, przy odpowiednich założeniach.
Podejście używane w tym samouczku można traktować jako kombinację technik używanych w SQD i kwantowej diagonalizacji Krylova (KQD). Połączone podejście jest czasami nazywane kwantową diagonalizacją Krylova opartą na próbkowaniu (SQKD). Zobacz Diagonalizację kwantową Krylova dla hamiltonianów sieciowych jako samouczek dotyczący metody KQD.
Ten samouczek jest oparty na pracy "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", do której można się odwołać po więcej szczegółów.
Jednodomieszkowy model Andersona (SIAM)
Jednowymiarowy hamiltonian SIAM jest sumą trzech wyrazów:
gdzie
Tutaj to fermionowe operatory kreacji/anihilacji dla miejsca kąpieli ze spinem , to operatory kreacji/anihilacji dla trybu domieszki, a . , i to liczby rzeczywiste opisujące odpowiednio oddziaływania przeskokowe, na miejscu i hybrydyzacyjne, zaś to liczba rzeczywista określająca potencjał chemiczny.
Zauważ, że hamiltonian jest szczególnym przypadkiem ogólnego hamiltoniana elektronów oddziałujących,
gdzie składa się z wyrazów jednociałowych, które są kwadratowe w fermionowych operatorach kreacji i anihilacji, a składa się z wyrazów dwuciałowych, które są czwartego stopnia. Dla SIAM,
a zawiera pozostałe wyrazy hamiltoniana. Aby reprezentować hamiltonian programowo, przechowujemy macierz i tensor .
Bazy położenia i pędu
Ze względu na przybliżoną symetrię translacyjną w , nie oczekujemy, że stan podstawowy będzie rzadki w bazie położenia (bazie orbitalnej, w której hamiltonian jest określony powyżej). Wydajność SQD jest gwarantowana tylko wtedy, gdy stan podstawowy jest rzadki, tzn. ma znaczące wagi tylko na małej liczbie stanów bazy obliczeniowej. Aby poprawić rzadkość stanu podstawowego, przeprowadzamy symulację w bazie orbitalnej, w której jest diagonalny. Tę bazę nazywamy bazą pędową. Ponieważ jest kwadratowym fermionowym hamiltonianem, może być efektywnie zdiagonalizowany przez obrót orbitalny.
Przybliżona ewolucja czasowa przez hamiltonian
Aby przybliżyć ewolucję czasową przez hamiltonian, używamy dekompozycji Trottera-Suzukiego drugiego rzędu,
W ramach transformacji Jordana-Wignera, ewolucja czasowa przez sprowadza się do pojedynczej bramki CPhase między orbitalami spin-góra i spin-dół w miejscu domieszki. Ponieważ jest kwadratowym fermionowym hamiltonianem, ewolucja czasowa przez sprowadza się do obrotu orbitalnego.
Stany bazowe Krylova , gdzie jest wymiarem podprzestrzeni Krylova, są tworzone przez wielokrotne zastosowanie pojedynczego kroku Trottera, więc
W poniższym przepływie pracy opartym na SQD będziemy próbkować z tego zestawu obwodów i post-procesować połączony zestaw bitstrings przy użyciu SQD. To podejście kontrastuje z tym stosowanym w powiązanym samouczku Kwantowa diagonalizacja oparta na próbkowaniu hamiltonowskiego chemicznego, gdzie próbki były pobierane z jednego heurystycznego wariacyjnego obwodu.
Wymagania
Przed rozpoczęciem tego samouczka upewnij się, że masz zainstalowane:
- Qiskit SDK v1.0 lub nowszy, z obsługą wizualizacji
- Qiskit Runtime v0.22 lub nowszy (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 lub nowszy (
pip install qiskit-addon-sqd) - ffsim v0.0.72 lub nowszy (
pip install ffsim)
Przykład na małą skalę z symulatorem
Krok 1: Odwzorowanie problemu na obwód kwantowy
Najpierw generujemy hamiltonian SIAM w bazie położenia. Hamiltonian jest reprezentowany przez macierz i tensor . Następnie obracamy go do bazy pędowej. W bazie położenia umieszczamy domieszkę na pierwszym miejscu. Jednak gdy obracamy do bazy pędowej, przesuwamy domieszkę na centralne miejsce, aby ułatwić oddziaływania z innymi orbitalami.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
Następnie generujemy obwody do produkcji stanów bazowych Krylova. Dla każdego gatunku spinowego, stan początkowy jest zadany przez superpozycję wszystkich możliwych wzbudzeń trzech elektronów najbliższych poziomu Fermiego do 4 najbliższych pustych trybów startując od stanu , realizowaną przez zastosowanie siedmiu bramek XXPlusYYGate. Stany po ewolucji czasowej są produkowane przez kolejne zastosowania kroku Trottera drugiego rzędu.
Bardziej szczegółowy opis tego modelu i sposobu projektowania obwodów znajduje się w pracy "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
Krok 2: Optymalizacja problemu pod kątem wykonania kwantowego
Następnie optymalizujemy obwód dla docelowego sprzętu. Na razie utworzymy generyczny Backend z określoną liczbą qubitów i zestawem bramek, do których obwody ewolucji czasowej naturalnie się rozkładają.
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
Teraz używamy Qiskit do transpilacji obwodów do docelowego backendu.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
Krok 3: Wykonanie przy użyciu prymitywów Qiskit
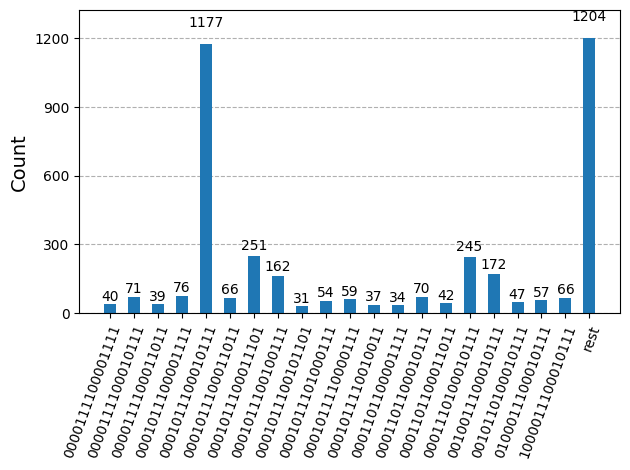
Po optymalizacji obwodów do wykonania na sprzęcie jesteśmy gotowi uruchomić je na docelowym sprzęcie i zebrać próbki do estymacji energii stanu podstawowego. Po użyciu prymitywu Sampler do próbkowania bitstrings z każdego obwodu łączymy wszystkie wyniki w jeden słownik zliczeń i rysujemy 20 najczęściej próbkowanych bitstrings.
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
Krok 4: Post-processing i zwrócenie wyniku do żądanego formatu klasycznego
Teraz uruchamiamy algorytm SQD przy użyciu funkcji diagonalize_fermionic_hamiltonian. Zobacz dokumentację API, aby zapoznać się z wyjaśnieniami argumentów tej funkcji.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
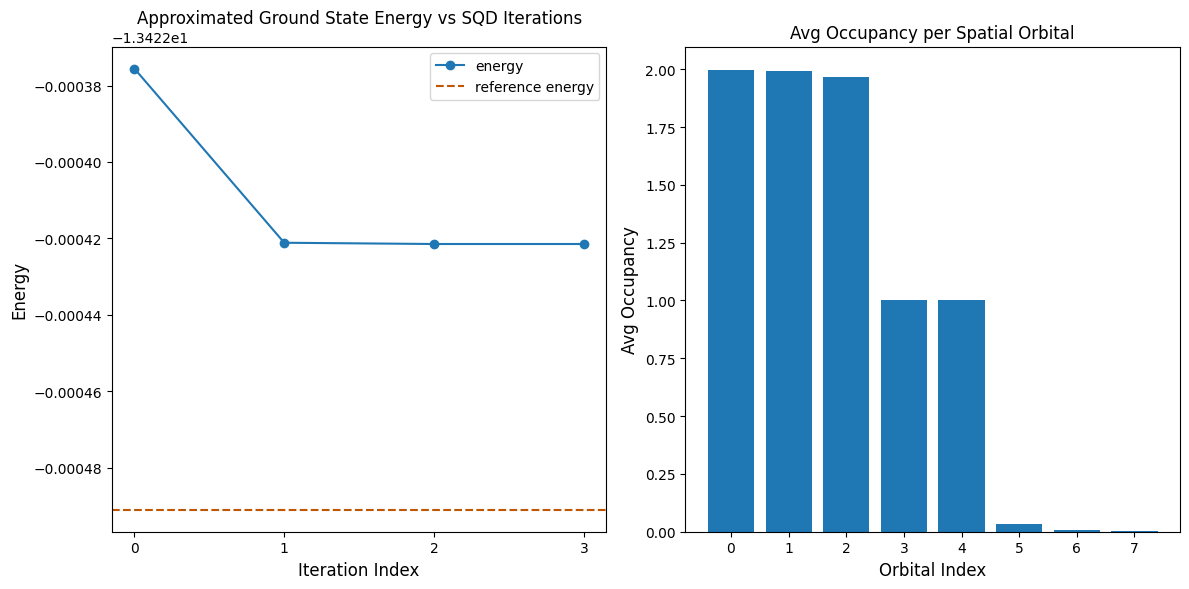
Poniższy fragment kodu rysuje wyniki. Pierwszy wykres pokazuje obliczoną energię w funkcji liczby iteracji odzyskiwania konfiguracji, a drugi wykres pokazuje średnie obsadzenie każdego orbitalu przestrzennego po ostatniej iteracji. Ponieważ jest to tak mały problem, już pierwsza iteracja przybliża nas bardzo blisko dokładnej energii (zwróć uwagę na skalę osi y).
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
Weryfikacja energii
Energia zwrócona przez SQD jest gwarantowana jako górne ograniczenie prawdziwej energii stanu podstawowego. Wartość energii można zweryfikować, ponieważ SQD zwraca również współczynniki wektora stanu przybliżającego stan podstawowy. Możesz obliczyć energię z wektora stanu przy użyciu jego jednocząsteczkowych i dwucząsteczkowych zredukowanych macierzy gęstości, jak pokazano w poniższym fragmencie kodu.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
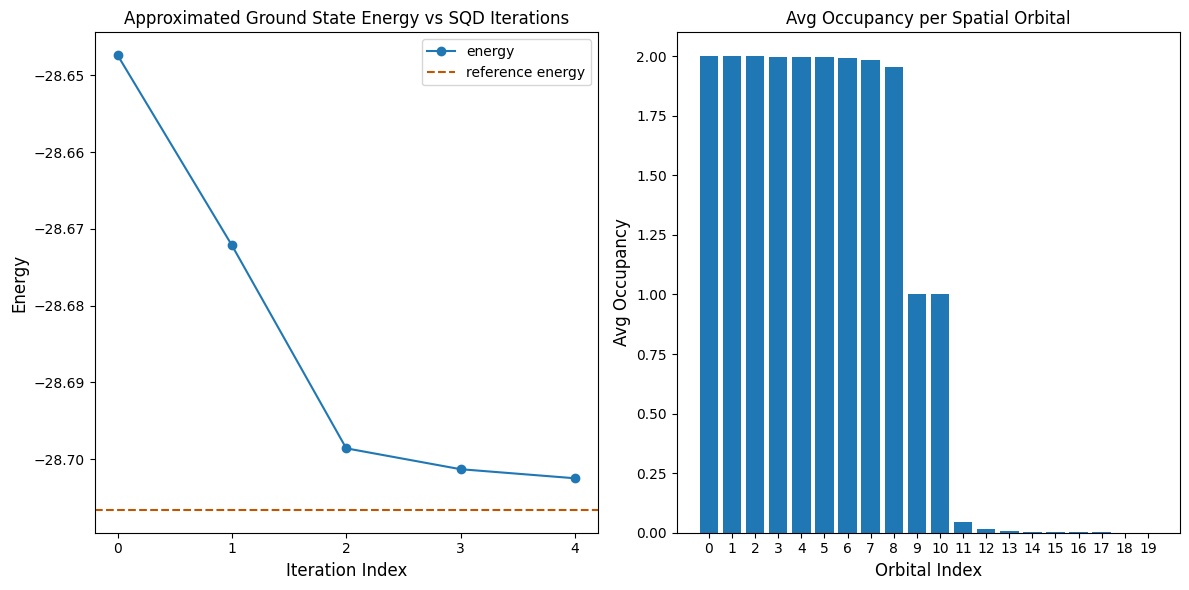
Przykład na dużą skalę na prawdziwym sprzęcie
Teraz uruchamiamy większy przykład na prawdziwym QPU. Jako energię referencyjną używamy wyników obliczenia DMRG przeprowadzonego osobno.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
Następne kroki
Jeśli ta praca Cię zainteresowała, możesz być zainteresowany następującymi materiałami:
- Kwantowa diagonalizacja oparta na próbkowaniu hamiltonowskiego chemicznego - powiązany samouczek używający heurystycznego wariacyjnego ansatzu zamiast obwodów Trottera
- Diagonalizacja kwantowa Krylova dla hamiltonianów sieciowych - samouczek dotyczący metody KQD
- Dokumentacja API dodatku SQD - odniesienie do funkcji
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization - artykuł, na którym oparty jest ten samouczek