Wariacyjny kwantowy solwer wartości własnych (VQE)
Ta lekcja wprowadza wariacyjny kwantowy solwer wartości własnych, wyjaśnia jego znaczenie jako fundamentalnego algorytmu w obliczeniach kwantowych, a także omawia jego mocne i słabe strony. VQE samo w sobie, bez metod uzupełniających, prawdopodobnie nie wystarczy do współczesnych obliczeń kwantowych na skalę użytkową. Mimo to jest ono ważne jako archetypiczna klasyczno-kwantowa metoda hybrydowa i stanowi istotny fundament, na którym budowanych jest wiele bardziej zaawansowanych algorytmów.
Ten film przedstawia ogólny zarys VQE oraz czynniki wpływające na jego wydajność. Tekst poniżej dodaje więcej szczegółów i implementuje VQE za pomocą Qiskit.
1. Czym jest VQE?
Wariacyjny kwantowy solwer wartości własnych to algorytm, który wykorzystuje obliczenia klasyczne i kwantowe łącznie, aby wykonać zadanie. W obliczeniu VQE występują 4 główne elementy:
- Operator: Często Hamiltonian, który będziemy oznaczać jako , opisujący właściwość systemu, którą chcesz zoptymalizować. Inaczej mówiąc, poszukujesz wektora własnego tego operatora odpowiadającego minimalnej wartości własnej. Często nazywamy ten wektor własny „stanem podstawowym".
- „Ansatz" (niemieckie słowo oznaczające „podejście"): jest to obwód kwantowy, który przygotowuje stan kwantowy przybliżający poszukiwany wektor własny. W rzeczywistości ansatz to rodzina obwodów kwantowych, ponieważ niektóre z bramek w ansatzu są sparametryzowane, to znaczy przyjmują parametr, który możemy zmieniać. Ta rodzina obwodów kwantowych może przygotować rodzinę stanów kwantowych przybliżających stan podstawowy.
- Estymator: środek do oszacowania wartości oczekiwanej operatora dla bieżącego wariacyjnego stanu kwantowego. Czasami naprawdę zależy nam po prostu na tej wartości oczekiwanej, którą nazywamy funkcją kosztu. Czasami interesuje nas bardziej skomplikowana funkcja, którą nadal można zapisać wychodząc od jednej lub więcej wartości oczekiwanych.
- Optymalizator klasyczny: algorytm, który zmienia parametry, próbując zminimalizować funkcję kosztu.
Przyjrzyjmy się każdemu z tych elementów bardziej szczegółowo.
1.1 Operator (Hamiltonian)
W rdzeniu problemu VQE znajduje się operator opisujący interesujący nas system. Zakładamy tutaj, że najmniejsza wartość własna i odpowiadający jej wektor własny tego operatora są użyteczne w jakimś celu naukowym lub biznesowym. Przykłady mogą obejmować Hamiltonian chemiczny opisujący cząsteczkę, taki że najmniejsza wartość własna operatora odpowiada energii stanu podstawowego cząsteczki, a odpowiadający jej stan własny opisuje geometrię lub konfigurację elektronową cząsteczki. Albo operator mógłby opisywać koszt pewnego procesu do zoptymalizowania, a stany własne mogłyby odpowiadać trasom lub praktykom. W niektórych dziedzinach, takich jak fizyka, „Hamiltonian" prawie zawsze odnosi się do operatora opisującego energię systemu fizycznego. Jednak w obliczeniach kwantowych powszechne jest spotykanie operatorów kwantowych, które opisują problem biznesowy lub logistyczny, również nazywanych „Hamiltonianem". Przyjmiemy tutaj tę konwencję.

Mapowanie problemu fizycznego lub optymalizacyjnego na kubity jest zazwyczaj nietrywialnym zadaniem, ale te szczegóły nie są głównym tematem tego kursu. Ogólną dyskusję na temat mapowania problemu na operator kwantowy można znaleźć w Kwantowe obliczenia w praktyce. Bardziej szczegółowe spojrzenie na mapowanie problemów chemicznych na operatory kwantowe można znaleźć w Chemia kwantowa z VQE.
Na potrzeby tego kursu założymy, że postać Hamiltonianu jest znana. Na przykład Hamiltonian dla prostej cząsteczki wodoru (przy pewnych założeniach dotyczących przestrzeni aktywnej oraz używając mappera Jordana-Wignera) to:
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy
from qiskit.quantum_info import SparsePauliOp
hamiltonian = SparsePauliOp(
[
"IIII",
"IIIZ",
"IZII",
"IIZI",
"ZIII",
"IZIZ",
"IIZZ",
"ZIIZ",
"IZZI",
"ZZII",
"ZIZI",
"YYYY",
"XXYY",
"YYXX",
"XXXX",
],
coeffs=[
-0.09820182 + 0.0j,
-0.1740751 + 0.0j,
-0.1740751 + 0.0j,
0.2242933 + 0.0j,
0.2242933 + 0.0j,
0.16891402 + 0.0j,
0.1210099 + 0.0j,
0.16631441 + 0.0j,
0.16631441 + 0.0j,
0.1210099 + 0.0j,
0.17504456 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
],
)
Zauważ, że w powyższym Hamiltonianie są takie wyrazy jak ZZII i YYYY, które nie komutują ze sobą. To znaczy, aby wyliczyć ZZII, musielibyśmy zmierzyć operator Pauli Z na kubicie 3 (wśród innych pomiarów). Ale aby wyliczyć YYYY, musimy zmierzyć operator Pauli Y na tym samym kubicie, kubicie 3. Istnieje relacja nieoznaczoności pomiędzy operatorami Y i Z na tym samym kubicie; nie możemy zmierzyć obu tych operatorów jednocześnie. Wrócimy do tego punktu poniżej, a właściwie przez cały kurs.
Powyższy Hamiltonian jest operatorem macierzowym . Diagonalizacja tego operatora w celu znalezienia jego minimalnej energetycznej wartości własnej nie jest trudna.
import numpy as np
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues), "hartrees")
The ground state energy is -1.1459778447627311 hartrees
Klasyczne solwery wartości własnych działające metodą brute force nie skalują się tak, aby opisać energie lub geometrie bardzo dużych układów atomów, takich jak leki czy białka. VQE jest jedną z wczesnych prób wykorzystania obliczeń kwantowych w tym problemie.
W tej lekcji napotkamy Hamiltoniany znacznie większe niż ten powyżej. Ale marnotrawstwem byłoby wypychanie granic tego, co VQE potrafi, zanim wprowadzimy niektóre z bardziej zaawansowanych narzędzi, które mogą uzupełnić lub zastąpić VQE, w dalszej części tego kursu.
1.2 Ansatz
Słowo „ansatz" pochodzi z języka niemieckiego i oznacza „podejście". Poprawną liczbą mnogą w języku niemieckim jest „ansätze", choć często można spotkać też formy „ansatzes" lub „ansatze". W kontekście VQE, ansatz to obwód kwantowy, którego używasz do utworzenia wielokubitowej funkcji falowej najbliżej przybliżającej stan podstawowy badanego układu, a zatem dającej najniższą wartość oczekiwaną twojego operatora. Ten obwód kwantowy będzie zawierał parametry wariacyjne (często zebrane razem w wektorze zmiennych ).

Wybierany jest początkowy zestaw wartości parametrów wariacyjnych. Operację unitarną ansatzu na obwodzie nazwiemy . Domyślnie wszystkie kubity w komputerach kwantowych IBM® są inicjalizowane do stanu . Po uruchomieniu obwodu stan kubitów będzie
Gdyby potrzebna była nam tylko najniższa energia (w języku układów fizycznych), moglibyśmy ją oszacować po prostu wielokrotnie mierząc energię i wybierając najniższą wartość. Ale zazwyczaj chcemy też poznać konfigurację, która daje tę najniższą energię lub wartość własną. Kolejnym krokiem jest więc oszacowanie wartości oczekiwanej Hamiltonianu, które jest dokonywane poprzez pomiary kwantowe. Dużo się na to składa. Możemy jednak zrozumieć ten proces jakościowo, zauważając, że prawdopodobieństwo zmierzenia energii (znów używając języka układów fizycznych) jest powiązane z wartością oczekiwaną wzorem:
Prawdopodobieństwo jest także powiązane z nakładaniem się stanu własnego i aktualnego stanu układu :
Tak więc, wykonując wiele pomiarów operatorów Pauli tworzących nasz Hamiltonian, możemy oszacować wartość oczekiwaną Hamiltonianu w aktualnym stanie układu . Następnym krokiem jest zmiana parametrów i próba bliższego podejścia do stanu o najniższej energii (stanu podstawowego) układu. Ze względu na parametry wariacyjne w ansatzu, czasami mówi się o nim jako o formie wariacyjnej.
Zanim przejdziemy do tego procesu wariacyjnego, zwróć uwagę, że często warto rozpocząć swój stan od „dobrze zgadniętego" stanu. Możesz wiedzieć wystarczająco dużo o swoim układzie, aby dokonać lepszego wstępnego przypuszczenia niż . Na przykład w zastosowaniach chemicznych często inicjalizuje się kubity do stanu Hartree-Focka. To początkowe przypuszczenie, które nie zawiera żadnych parametrów wariacyjnych, nazywa się stanem referencyjnym. Nazwijmy obwód kwantowy używany do utworzenia stanu referencyjnego . Gdy ważne staje się rozróżnienie stanu referencyjnego od reszty ansatzu, używamy: Równoważnie
1.3 Estimator
Potrzebujemy sposobu na oszacowanie wartości oczekiwanej naszego Hamiltonianu w konkretnym stanie wariacyjnym . Gdybyśmy mogli bezpośrednio zmierzyć cały operator , byłoby to tak proste, jak wykonanie wielu (powiedzmy ) pomiarów i uśrednienie zmierzonych wartości:
Tutaj symbol przypomina nam, że ta wartość oczekiwana byłaby dokładnie poprawna tylko w granicy . Ale przy wykonywaniu tysięcy pomiarów na obwodzie błąd próbkowania wartości oczekiwanej jest dość niski. Istnieją inne kwestie, takie jak szum, które stają się problemem przy bardzo precyzyjnych obliczeniach.
Jednak zwykle nie jest możliwe zmierzenie całego naraz. może zawierać wiele nieprzemiennych operatorów Pauli X, Y i Z. Hamiltonian musi więc zostać rozbity na grupy operatorów, które można mierzyć jednocześnie, a każda taka grupa musi być oszacowana osobno, po czym wyniki łączy się w celu otrzymania wartości oczekiwanej. Wrócimy do tego bardziej szczegółowo w następnej lekcji, gdy omówimy skalowanie podejść klasycznych i kwantowych. Ta złożoność w pomiarach jest jednym z powodów, dla których potrzebujemy wysoce wydajnego kodu do przeprowadzania takich estymacji. W tej i kolejnych lekcjach będziemy w tym celu korzystać z prymitywu Qiskit Runtime Estimator.
1.4 Klasyczne optymalizatory
Klasyczny optymalizator to dowolny klasyczny algorytm zaprojektowany do znajdowania ekstremów funkcji docelowej (zazwyczaj minimum). Przeszukują one przestrzeń możliwych parametrów w poszukiwaniu zestawu minimalizującego pewną interesującą nas funkcję. Można je ogólnie podzielić na metody gradientowe, które wykorzystują informację o gradiencie, oraz metody bezgradientowe, które działają jako optymalizatory typu black-box. Wybór klasycznego optymalizatora może znacząco wpłynąć na wydajność algorytmu, zwłaszcza w obecności szumu sprzętu kwantowego. Popularne optymalizatory w tej dziedzinie to Adam, AMSGrad i SPSA, które dały obiecujące wyniki w środowiskach zaszumionych. Bardziej tradycyjne optymalizatory to COBYLA i SLSQP.
Typowy przepływ pracy (pokazany w sekcji 3.3) polega na używaniu jednego z tych algorytmów jako metody wewnątrz minimalizatora, takiego jak funkcja minimize z scipy. Przyjmuje ona jako argumenty:
- Pewną funkcję do zminimalizowania. Często jest to wartość oczekiwana energii. Jednak ogólnie nazywa się je „funkcjami kosztu".
- Zestaw parametrów, od których rozpocząć przeszukiwanie. Często nazywany lub .
- Argumenty, w tym argumenty funkcji kosztu. W obliczeniach kwantowych z Qiskit, argumenty te obejmą ansatz, Hamiltonian i estymator, o czym szerzej w następnej podsekcji.
- „Metodę" (method) minimalizacji. Odnosi się to do konkretnego algorytmu używanego do przeszukiwania przestrzeni parametrów. Tutaj wskazalibyśmy na przykład COBYLA lub SLSQP.
- Opcje. Dostępne opcje mogą się różnić w zależności od metody. Ale przykładem, który praktycznie wszystkie metody by zawierały, jest maksymalna liczba iteracji optymalizatora przed zakończeniem przeszukiwania: 'maxiter'.

Na każdym kroku iteracyjnym wartość oczekiwana Hamiltonianu jest szacowana poprzez wykonanie wielu pomiarów. Ta oszacowana energia jest zwracana przez funkcję kosztu, a minimalizator aktualizuje informacje, jakie posiada o krajobrazie energii. To, co dokładnie optymalizator robi, aby wybrać następny krok, różni się w zależności od metody. Niektóre używają gradientów i wybierają kierunek najszybszego spadku. Inne mogą brać pod uwagę szum i wymagać, aby koszt zmniejszył się o dużą wielkość, zanim zaakceptują, że prawdziwa energia maleje w tym kierunku.
# Example syntax for minimization
# from scipy.optimize import minimize
# res = minimize(cost_func, x0, args=(ansatz, hamiltonian, estimator), method="cobyla",
# options={'maxiter': 200})
1.5 Zasada wariacyjna
W tym kontekście bardzo ważna jest zasada wariacyjna; stwierdza ona, że żadna wariacyjna funkcja falowa nie może dać wartości oczekiwanej energii (lub kosztu) niższej niż ta dawana przez funkcję falową stanu podstawowego. Matematycznie,
Łatwo to zweryfikować, zauważając, że zbiór wszystkich stanów własnych operatora tworzy kompletną bazę przestrzeni Hilberta. Innymi słowy, dowolny stan, a w szczególności , może zostać zapisany jako ważona (znormalizowana) suma tych stanów własnych :
gdzie są stałymi do wyznaczenia, a . Pozostawiamy to jako ćwiczenie dla czytelnika. Zwróć jednak uwagę na implikację: stan wariacyjny dający najniższą wartość oczekiwaną energii jest najlepszym oszacowaniem prawdziwego stanu podstawowego.
Sprawdź swoje zrozumienie
Przeczytaj poniższe pytanie, pomyśl o odpowiedzi, a następnie kliknij trójkąt, aby odsłonić rozwiązanie.
Zweryfikuj matematycznie, że dla dowolnego stanu wariacyjnego .
Odpowiedź:
Korzystając z danego rozwinięcia stanu wariacyjnego w stanach własnych energii,
możemy zapisać wariacyjną wartość oczekiwaną energii jako
Dla wszystkich współczynników . Możemy więc zapisać
2. Porównanie z klasycznym przepływem pracy
Załóżmy, że interesuje nas macierz o N wierszach i N kolumnach. Przypuśćmy, że macierz jest tak duża, iż dokładna diagonalizacja nie wchodzi w grę. Załóżmy dalej, że wiesz wystarczająco dużo o swoim problemie, aby móc poczynić pewne przypuszczenia co do ogólnej struktury docelowego stanu własnego, i chcesz zbadać stany podobne do swojego początkowego przypuszczenia, aby zobaczyć, czy można dalej obniżyć koszt/energię. To podejście wariacyjne i jest jedną z metod stosowanych, gdy dokładna diagonalizacja nie wchodzi w grę.
2.1 Klasyczny przepływ pracy
Przy użyciu klasycznego komputera wyglądałoby to następująco:
- Wykonaj stan będący przypuszczeniem, z pewnymi parametrami , które będziesz zmieniać: . Chociaż to początkowe przypuszczenie mogłoby być losowe, nie jest to zalecane. Chcemy wykorzystać wiedzę o rozważanym problemie, aby dostosować nasze przypuszczenie najlepiej jak to możliwe.
- Oblicz wartość oczekiwaną operatora dla układu w tym stanie:
- Zmień parametry wariacyjne i powtórz: .
- Wykorzystaj zgromadzoną informację o krajobrazie możliwych stanów w Twojej podprzestrzeni wariacyjnej, aby tworzyć coraz lepsze przypuszczenia i zbliżać się do stanu docelowego. Zasada wariacyjna gwarantuje, że nasz stan wariacyjny nie może dać wartości własnej niższej niż stan podstawowy docelowy. Zatem im niższa wartość oczekiwana, tym lepsze nasze przybliżenie stanu podstawowego:
Przeanalizujmy trudność każdego kroku w tym podejściu. Ustawianie lub aktualizowanie parametrów jest łatwe obliczeniowo; trudność polega tu na wybraniu użytecznych, fizycznie umotywowanych parametrów początkowych. Wykorzystanie zgromadzonej informacji z poprzednich iteracji do aktualizowania parametrów w taki sposób, aby zbliżać się do stanu podstawowego, jest nietrywialne. Jednak istnieją klasyczne algorytmy optymalizacji, które robią to całkiem efektywnie. Ta klasyczna optymalizacja jest kosztowna tylko dlatego, że może wymagać wielu iteracji; w najgorszym przypadku liczba iteracji może skalować się wykładniczo z N. Najbardziej kosztownym obliczeniowo pojedynczym krokiem jest prawie na pewno obliczenie wartości oczekiwanej Twojej macierzy przy użyciu danego stanu :
Macierz musi działać na wektor o elementach, co odpowiada: operacjom mnożenia w najgorszym przypadku. Musi to być wykonane w każdej iteracji parametrów. Dla niezwykle dużych macierzy wiąże się to z wysokim kosztem obliczeniowym.
2.2 Kwantowy przepływ pracy i komutujące grupy Pauli
Teraz wyobraź sobie przekazanie tej części obliczeń komputerowi kwantowemu. Zamiast obliczać tę wartość oczekiwaną, szacujesz ją, przygotowując stan na komputerze kwantowym za pomocą swojego ansatzu wariacyjnego, a następnie wykonując pomiary.
Może to brzmieć łatwiej niż jest w rzeczywistości. Ogólnie nie jest łatwy do zmierzenia. Na przykład może składać się z wielu niekomutujących operatorów Pauli X, Y i Z. Ale może być zapisany jako kombinacja liniowa wyrazów , z których każdy jest łatwo mierzalny (na przykład operatory Pauli lub grupy komutujących kubitowo operatorów Pauli). Wartość oczekiwana w pewnym stanie jest ważoną sumą wartości oczekiwanych wyrazów składowych . Wyrażenie to obowiązuje dla dowolnego stanu , ale będziemy je stosować szczególnie do naszych stanów wariacyjnych .
gdzie jest ciągiem Pauli jak IZZX…XIYX, lub kilkoma takimi ciągami, które komutują ze sobą. Zatem opis wartości oczekiwanej bliższy realiom pomiarów na komputerach kwantowych to
A w kontekście naszej wariacyjnej funkcji falowej:
Każdy z wyrazów może być mierzony razy, dając próbki pomiarowe z i zwraca wartość oczekiwaną oraz odchylenie standardowe . Możemy sumować te wyrazy i propagować błędy przez sumę, aby uzyskać ogólną wartość oczekiwaną i odchylenie standardowe .
Nie wymaga to mnożenia na dużą skalę ani żadnego procesu, który musiałby skalować się jak . Zamiast tego wymaga wielu pomiarów na komputerze kwantowym. Jeśli nie potrzebujesz zbyt wielu takich pomiarów, to podejście może być efektywne. I to jest kwantowa część VQE.
Ale porozmawiajmy o powodach, dla których może to nie być efektywne. Jednym z powodów wielu pomiarów jest zmniejszenie statystycznej niepewności Twoich oszacowań, dla obliczeń o bardzo wysokiej precyzji. Innym powodem jest liczba ciągów Pauli wymaganych do rozpięcia całej Twojej macierzy. Ponieważ macierze Pauli (plus identyczność: X, Y, Z oraz I) rozpinają przestrzeń wszystkich operatorów danego wymiaru, mamy gwarancję, że możemy zapisać interesującą nas macierz jako ważoną sumę operatorów Pauli, jak zrobiliśmy wcześniej.
gdzie jest ciągiem Pauli działającym na wszystkich kubitach opisujących Twój układ, jak IZZX…XIYX, lub kilkoma takimi ciągami, które komutują ze sobą. Przypomnijmy, że Qiskit używa notacji little endian, w której operator Pauli od prawej działa na kubit. Możemy więc zmierzyć nasz operator poprzez zmierzenie serii operatorów Pauli.
Ale nie możemy mierzyć wszystkich tych operatorów Pauli jednocześnie. Operatory Pauli (z wyjątkiem I) nie komutują ze sobą, jeśli są związane z tym samym kubitem. Na przykład możemy mierzyć IZIZ i ZZXZ jednocześnie, ponieważ możemy mierzyć I i Z jednocześnie dla trzeciego kubita, a także możemy znać I i X jednocześnie dla pierwszego kubita. Ale nie możemy mierzyć ZZZZ i ZZZX jednocześnie, ponieważ Z i X nie komutują, a oba działają na 0. kubit. Doświadczeni czytelnicy mogą pamiętać, że dwie grupy operatorów Pauli mogą komutować jako zbiór, nawet jeśli pomiary poszczególnych kubitów nie komutują. Estimator zakłada pomiary Pauliego w postaci iloczynu tensorowego (poprzez rotacje bazowe), co odpowiada grupowaniu operatorów komutujących kubitowo. Aby jednocześnie oszacować dwa ciągi (A i B) operatorów Pauli za pomocą Estimatora, operatory Pauliego każdego kubita w A i B muszą komutować. Oznacza to, że nie możemy też mierzyć ZZZZ i ZZXX jednocześnie.

Dekomponujemy więc naszą macierz na sumę operatorów Pauli działających na różne kubity. Niektóre elementy tej sumy mogą być mierzone wszystkie naraz; nazywamy to grupą komutujących operatorów Pauli. W zależności od tego, ile jest wyrazów niekomutujących, możemy potrzebować wielu takich grup. Oznaczmy liczbę takich grup komutujących ciągów Pauli jako . Jeśli jest małe, może to działać dobrze. Jeśli ma miliony grup, nie będzie to użyteczne.
Procesy wymagane do oszacowania wartości oczekiwanej są zebrane razem w prymitywie Qiskit Runtime o nazwie Estimator. Aby dowiedzieć się więcej o Estimator, zobacz API reference w dokumentacji IBM Quantum®. Można po prostu użyć Estimator bezpośrednio, ale Estimator zwraca znacznie więcej niż tylko najniższą wartość własną energii. Na przykład zwraca także informacje o błędzie standardowym zespołu. Dlatego w kontekście problemów minimalizacji często widzi się Estimator wewnątrz funkcji kosztu. Aby dowiedzieć się więcej o wejściach i wyjściach Estimator, zobacz ten przewodnik w dokumentacji IBM Quantum.
Zapisujesz wartość oczekiwaną (lub funkcję kosztu) dla zestawu parametrów użytych w Twoim stanie, a następnie aktualizujesz parametry. Z czasem możesz używać wartości oczekiwanych lub wartości funkcji kosztu, które oszacowałeś, aby przybliżyć gradient funkcji kosztu w podprzestrzeni stanów próbkowanych przez Twój ansatz. Istnieją zarówno klasyczne optymalizatory oparte na gradiencie, jak i bez gradientu. Oba cierpią na potencjalne problemy z trenowalnością, takie jak wiele lokalnych minimów oraz duże obszary przestrzeni parametrów z gradientem bliskim zeru, zwane jałowymi płaskowyżami.

2.3 Czynniki determinujące koszt obliczeniowy
VQE nie rozwiąże wszystkich Twoich najtrudniejszych problemów chemii kwantowej. Nie. Ale bycie lepszym we wszystkich obliczeniach nie jest celem. Przesunęliśmy to, co determinuje koszt obliczeniowy.

Przeszliśmy od procesu, którego złożoność zależy tylko od wymiaru macierzy, do takiego, który zależy od wymaganej precyzji i liczby niekomutujących operatorów Pauli, z których składa się macierz. Ta ostatnia kwestia nie ma odpowiednika w klasycznych obliczeniach.
Na podstawie tych zależności, dla macierzy rzadkich lub macierzy obejmujących niewiele niekomutujących ciągów Pauli, ten proces może być użyteczny. Tak jest na przykład w przypadku układów oddziałujących spinów. Dla macierzy gęstych może być mniej użyteczny. Wiemy na przykład, że układy chemiczne często mają Hamiltoniany obejmujące setki, tysiące, a nawet miliony ciągów Pauli. Wykonano interesującą pracę nad zmniejszeniem tej liczby wyrazów. Ale układy chemiczne mogą być lepiej dopasowane do niektórych innych algorytmów, które omówimy na tym kursie.
Sprawdź swoje zrozumienie
Przeczytaj poniższe pytania, zastanów się nad odpowiedziami, a następnie kliknij trójkąty, aby ujawnić rozwiązania.
Rozważ Hamiltonian na czterech kubitach, który zawiera wyrazy:
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXX
Chcesz posortować te wyrazy w grupy w taki sposób, aby wszystkie wyrazy w grupie mogły być mierzone jednocześnie. Jaka jest najmniejsza liczba takich grup, które możesz utworzyć, aby wszystkie wyrazy zostały uwzględnione?
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXXOdpowiedź:
Można to zrobić w 4 grupach. Zauważ, że takie rozwiązania zazwyczaj nie są jednoznaczne.
IIXX, XXXX, IIZZ, ZZZZ
IIXZ, IZXZ, ZIXZ, ZZXZ
IXXZ
XZXZ
Co Twoim zdaniem zazwyczaj sprawia, że chemia kwantowa z VQE jest trudna: liczba wyrazów w Hamiltonianie czy znalezienie dobrego ansatzu?
Odpowiedź:
Okazuje się, że istnieją ansätze wysoce zoptymalizowane dla kontekstów chemicznych. Liczba wyrazów w Hamiltonianie, a co za tym idzie liczba wymaganych pomiarów, zazwyczaj powoduje więcej problemów.
3. Przykładowy Hamiltonian
Zastosujmy ten algorytm w praktyce, używając niewielkiej macierzy Hamiltonianu, aby zobaczyć, co dzieje się na każdym kroku. Skorzystamy z frameworka wzorców Qiskit (Qiskit patterns):
-Krok 1: Odwzorowanie problemu na obwody kwantowe i operatory -Krok 2: Optymalizacja pod docelowy sprzęt -Krok 3: Uruchomienie na docelowym sprzęcie -Krok 4: Przetwarzanie końcowe wyników
3.1 Krok 1: Odwzorowanie problemu na obwody kwantowe i operatory
Użyjemy tego zdefiniowanego powyżej w kontekście chemicznym. Zaczynamy od kilku ogólnych importów.
# General imports
import numpy as np
# SciPy minimizer routine
from scipy.optimize import minimize
# Plotting functions
import matplotlib.pyplot as plt
Ponownie zakładamy, że interesujący nas Hamiltonian jest znany. Użyjemy tutaj bardzo małego Hamiltonianu, ponieważ inne metody omawiane w tym kursie będą bardziej efektywne przy rozwiązywaniu większych problemów.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
hamiltonian = SparsePauliOp.from_list(
[("YZ", 0.3980), ("ZI", -0.3980), ("ZZ", -0.0113), ("XX", 0.1810)]
)
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues))
The ground state energy is -0.702930394459531
W Qiskit dostępnych jest wiele gotowych ansatzów. Użyjemy efficient_su2.
# Pre-defined ansatz circuit and operator class for Hamiltonian
from qiskit.circuit.library import efficient_su2
# Note that it is more common to place initial 'h' gates outside the ansatz.
# Here we specifically wanted this layer structure.
ansatz = efficient_su2(
hamiltonian.num_qubits, su2_gates=["h", "rz", "y"], entanglement="circular", reps=1
)
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.decompose().draw("mpl", style="iqp")
This circuit has 4 parameters
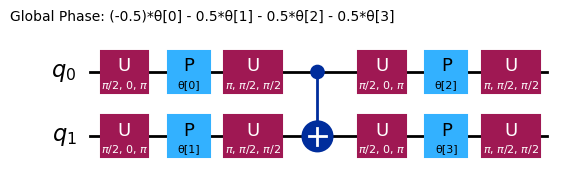
Różne ansatze mają różne struktury splątujące i różne bramki rotacyjne. Ten przedstawiony tutaj wykorzystuje bramki CNOT do splątywania, oraz zarówno bramki Y, jak i parametryzowane bramki RZ do rotacji. Zwróć uwagę na rozmiar tej przestrzeni parametrów; oznacza to, że musimy zminimalizować funkcję kosztu po 4 zmiennych (parametrach dla bramek RZ). Można to skalować, ale nie w nieskończoność. Uruchomienie podobnego problemu na 4 kubitach, przy domyślnych 3 powtórzeniach dla efficient_su2, daje 16 parametrów wariacyjnych.
3.2 Krok 2: Optymalizacja pod docelowy sprzęt
Ansatz został zapisany z użyciem znanych bramek, ale nasz obwód musi zostać przetranspilowany, aby wykorzystać bramki bazowe, które można zaimplementować na każdym komputerze kwantowym. Wybieramy najmniej obciążony backend.
# runtime imports
from qiskit_ibm_runtime import QiskitRuntimeService, Session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# To run on hardware, select the backend with the fewest number of jobs in the queue
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
print(backend)
<IBMBackend('ibm_torino')>
Teraz możemy przetranspilować nasz obwód pod ten sprzęt i zwizualizować przetranspilowany ansatz.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")

Zwróć uwagę, że użyte bramki uległy zmianie, a kubity w naszym abstrakcyjnym obwodzie zostały odwzorowane na inaczej ponumerowane kubity w komputerze kwantowym. Musimy odwzorować nasz Hamiltonian identycznie, aby nasze wyniki były znaczące.
hamiltonian_isa = hamiltonian.apply_layout(layout=ansatz_isa.layout)
3.3 Krok 3: Uruchomienie na docelowym sprzęcie
3.3.1 Raportowanie wartości
Definiujemy tutaj funkcję kosztu, która przyjmuje jako argumenty struktury zbudowane w poprzednich krokach: parametry, ansatz oraz Hamiltonian. Korzysta ona także z estymatora (Estimator), którego jeszcze nie zdefiniowaliśmy. Dołączamy kod śledzący historię funkcji kosztu, abyśmy mogli sprawdzić zachowanie zbieżności.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from Estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
Bardzo korzystnie jest móc dobrać wartości początkowe parametrów na podstawie wiedzy o rozważanym problemie i charakterystyce stanu docelowego. Nie będziemy zakładać takiej wiedzy i użyjemy losowych wartości początkowych.
x0 = 2 * np.pi * np.random.random(num_params)
# This required 13 min, 20 s QPU time on an Eagle processor, 28 min total time.
with Session(backend=backend) as session:
estimator = Estimator(mode=session)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 50},
)
Iters. done: 1 [Current cost: 0.010575798722044727]
Iters. done: 2 [Current cost: 0.004040015974440895]
Iters. done: 3 [Current cost: 0.0020213258785942503]
Iters. done: 4 [Current cost: 0.18723082446726014]
Iters. done: 5 [Current cost: -0.2746792152068885]
Iters. done: 6 [Current cost: -0.3094547651648519]
Iters. done: 7 [Current cost: -0.05281985428356641]
Iters. done: 8 [Current cost: 0.00808560303514377]
Iters. done: 9 [Current cost: -0.0014821685303514388]
Iters. done: 10 [Current cost: -0.004759824281150161]
Iters. done: 11 [Current cost: 0.09942328705995292]
Iters. done: 12 [Current cost: 0.01092366214057508]
Iters. done: 13 [Current cost: 0.05017497496069776]
Iters. done: 14 [Current cost: 0.13028868414310696]
Iters. done: 15 [Current cost: 0.013747803514376994]
Iters. done: 16 [Current cost: 0.2583072432944498]
Iters. done: 17 [Current cost: -0.14422125655131562]
Iters. done: 18 [Current cost: -0.0004950150347678081]
Iters. done: 19 [Current cost: 0.00681082268370607]
Iters. done: 20 [Current cost: -0.0023377795527156544]
Iters. done: 21 [Current cost: 0.6027665591169237]
Iters. done: 22 [Current cost: 0.00596641373801917]
Iters. done: 23 [Current cost: -0.008318769968051117]
Iters. done: 24 [Current cost: -0.00026683306709265246]
Iters. done: 25 [Current cost: -0.007648222843450479]
Iters. done: 26 [Current cost: 0.004121086261980831]
Iters. done: 27 [Current cost: -0.004075019968051117]
Iters. done: 28 [Current cost: -0.004419369009584665]
Iters. done: 29 [Current cost: 0.213185460054037]
Iters. done: 30 [Current cost: -0.06505919572162797]
Iters. done: 31 [Current cost: -0.5334241316590271]
Iters. done: 32 [Current cost: 0.00218370607028754]
Iters. done: 33 [Current cost: 0.09579352143666908]
Iters. done: 34 [Current cost: -0.009274800319488819]
Iters. done: 35 [Current cost: -0.44395141360688106]
Iters. done: 36 [Current cost: 0.011747104632587858]
Iters. done: 37 [Current cost: -0.003344149361022364]
Iters. done: 38 [Current cost: 0.19138183916486304]
Iters. done: 39 [Current cost: 0.013513931813145209]
Możemy spojrzeć na surowe wyniki.
res
message: Return from COBYLA because the trust region radius reaches its lower bound.
success: True
status: 0
fun: -0.5334241316590271
x: [ 1.024e+00 6.459e+00 3.625e+00 4.007e+00]
nfev: 39
maxcv: 0.0
3.4 Krok 4: Przetwarzanie końcowe wyników
Jeśli procedura zakończy się poprawnie, wartości w naszym słowniku powinny być równe odpowiednio wektorowi rozwiązania i całkowitej liczbie ewaluacji funkcji. Łatwo to zweryfikować:
cost_history_dict
{'prev_vector': array([1.02397956, 6.45886604, 3.62479262, 4.00744128]),
'iters': 39,
'cost_history': [np.float64(0.010575798722044727),
np.float64(0.004040015974440895),
np.float64(0.0020213258785942503),
np.float64(0.18723082446726014),
np.float64(-0.2746792152068885),
np.float64(-0.3094547651648519),
np.float64(-0.05281985428356641),
np.float64(0.00808560303514377),
np.float64(-0.0014821685303514388),
np.float64(-0.004759824281150161),
np.float64(0.09942328705995292),
np.float64(0.01092366214057508),
np.float64(0.05017497496069776),
np.float64(0.13028868414310696),
np.float64(0.013747803514376994),
np.float64(0.2583072432944498),
np.float64(-0.14422125655131562),
np.float64(-0.0004950150347678081),
np.float64(0.00681082268370607),
np.float64(-0.0023377795527156544),
np.float64(0.6027665591169237),
np.float64(0.00596641373801917),
np.float64(-0.008318769968051117),
np.float64(-0.00026683306709265246),
np.float64(-0.007648222843450479),
np.float64(0.004121086261980831),
np.float64(-0.004075019968051117),
np.float64(-0.004419369009584665),
np.float64(0.213185460054037),
np.float64(-0.06505919572162797),
np.float64(-0.5334241316590271),
np.float64(0.00218370607028754),
np.float64(0.09579352143666908),
np.float64(-0.009274800319488819),
np.float64(-0.44395141360688106),
np.float64(0.011747104632587858),
np.float64(-0.003344149361022364),
np.float64(0.19138183916486304),
np.float64(0.013513931813145209)]}
fig, ax = plt.subplots()
x = np.linspace(0, 10, 50)
# Define the constant function
constant = -0.7029
y_constant = np.full_like(x, constant)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

IBM Quantum oferuje również inne materiały do podnoszenia kwalifikacji związane z VQE. Jeśli jesteś gotowy zastosować VQE w praktyce, zobacz nasz samouczek: Estymacja energii stanu podstawowego łańcucha Heisenberga za pomocą VQE. Jeśli chcesz uzyskać więcej informacji na temat tworzenia Hamiltonianów molekularnych, zobacz tę lekcję w naszym kursie Chemia kwantowa z VQE. Jeśli interesuje cię głębsze zrozumienie tego, jak działają algorytmy wariacyjne, takie jak VQE, polecamy kurs Projektowanie algorytmów wariacyjnych.
Sprawdź swoje zrozumienie
Przeczytaj poniższe pytanie, zastanów się nad odpowiedzią, a następnie kliknij trójkąt, aby ujawnić rozwiązanie.
W tej sekcji obliczyliśmy energię stanu podstawowego na podstawie Hamiltonianu. Gdybyśmy chcieli zastosować to na przykład do wyznaczania geometrii cząsteczki, jak moglibyśmy to rozszerzyć?
Odpowiedź:
Musielibyśmy wprowadzić zmienne dla odległości międzyatomowych oraz kątów między wiązaniami. Musielibyśmy je zmieniać. Dla każdej wariacji tych zmiennych generowalibyśmy nowy Hamiltonian (ponieważ operatory opisujące energię z pewnością zależą od geometrii). Dla każdego takiego wytworzonego Hamiltonianu odwzorowanego na kubity musielibyśmy przeprowadzić optymalizację podobną do tej wykonanej powyżej. Spośród wszystkich tych wielu zbieżnych problemów optymalizacyjnych, geometria, która dała najniższą energię, byłaby tą przyjętą przez naturę. Jest to znacznie bardziej skomplikowane niż to, co pokazano powyżej. Takie obliczenia są wykonywane dla najprostszej cząsteczki, , tutaj.
4. Związek VQE z innymi metodami
W tej sekcji przyjrzymy się zaletom i wadom oryginalnego podejścia VQE oraz wskażemy jego powiązania z innymi, nowszymi algorytmami.
4.1 Mocne i słabe strony VQE
Niektóre mocne strony zostały już wskazane. Należą do nich:
- Przystosowanie do nowoczesnego sprzętu: Niektóre algorytmy kwantowe wymagają znacznie niższych wskaźników błędów, zbliżających się do tolerancji błędów w dużej skali. VQE nie ma takich wymagań; można go zaimplementować na obecnych komputerach kwantowych.
- Płytkie obwody: VQE często wykorzystuje stosunkowo płytkie obwody kwantowe. Dzięki temu VQE jest mniej podatny na skumulowane błędy bramek i nadaje się do wielu technik łagodzenia błędów. Oczywiście obwody nie zawsze są płytkie; zależy to od zastosowanego ansatzu.
- Wszechstronność: VQE można (w zasadzie) zastosować do każdego problemu, który można sformułować jako problem wartości/wektorów własnych. Istnieje wiele zastrzeżeń, które sprawiają, że VQE jest niepraktyczny lub niekorzystny dla niektórych problemów. Niektóre z nich podsumowano poniżej.
Powyżej opisano również pewne słabości VQE oraz problemy, dla których jest on niepraktyczny. Należą do nich:
- Heurystyczny charakter: VQE nie gwarantuje zbieżności do prawidłowej energii stanu podstawowego, ponieważ jego wydajność zależy od wyboru ansatzu i metod optymalizacji[1-2]. Jeśli wybrany zostanie słaby ansatz, któremu brakuje wymaganego splątania dla pożądanego stanu podstawowego, żaden klasyczny optymalizator nie będzie w stanie osiągnąć tego stanu podstawowego.
- Potencjalnie liczne parametry: Bardzo ekspresyjny ansatz może mieć tak wiele parametrów, że iteracje minimalizacji są bardzo czasochłonne.
- Wysoki narzut pomiarowy: W VQE estymator służy do oszacowania wartości oczekiwanej każdego wyrazu w Hamiltonianie. Większość interesujących nas Hamiltonianów będzie miała wyrazy, których nie można jednocześnie oszacować. To może sprawić, że VQE będzie zasobochłonny dla dużych układów ze skomplikowanymi Hamiltonianami[1].
- Wpływ szumu: Gdy klasyczny optymalizator szuka minimum, zaszumione obliczenia mogą go zdezorientować i skierować z dala od prawdziwego minimum lub opóźnić jego zbieżność. Jednym z możliwych rozwiązań jest wykorzystanie najnowocześniejszych technik łagodzenia błędów i tłumienia błędów[2-3] firmy IBM.
- Jałowe płaskowyże: Te obszary zanikających gradientów[2-3] istnieją nawet przy braku szumu, ale szum czyni je bardziej problematycznymi, ponieważ zmiana wartości oczekiwanych z powodu szumu może być większa niż zmiana spowodowana aktualizacją parametrów w tych jałowych obszarach.
4.2 Związek z innymi podejściami
Adapt-VQE
Algorytm ADAPT-VQE (Adaptive Derivative-Assembled Pseudo-Trotter Variational Quantum Eigensolver) jest ulepszeniem oryginalnego algorytmu VQE, zaprojektowanym w celu poprawy wydajności, dokładności i skalowalności dla symulacji kwantowych, szczególnie w chemii kwantowej.
Oryginalny algorytm VQE opisany w tej lekcji wykorzystuje predefiniowany, stały ansatz do aproksymacji stanu podstawowego układu. W naszym przypadku użyliśmy efficient_su2, z pojedynczym powtórzeniem, używając bramek rotacji Y i RZ. Chociaż parametry w bramkach RZ się zmieniały, struktura tego ansatzu i zastosowane bramki nie uległy zmianie.
ADAPT-VQE eliminuje ograniczenia VQE poprzez adaptacyjną konstrukcję ansatzu. Zamiast rozpoczynać od stałego ansatzu, ADAPT-VQE dynamicznie buduje ansatz iteracyjnie. W każdym kroku wybiera z predefiniowanej puli (takiej jak operatory wzbudzenia fermionowego) ten operator, który ma największy gradient względem energii. Dzięki temu dodawane są tylko najbardziej wpływowe operatory, co prowadzi do kompaktowego i wydajnego ansatzu[4-6]. Takie podejście może mieć kilka korzystnych skutków:
- Zmniejszona głębokość obwodu: Poprzez stopniowe rozbudowywanie ansatzu i skupianie się tylko na niezbędnych operatorach, ADAPT-VQE minimalizuje operacje bramek w porównaniu z tradycyjnymi podejściami VQE[5,7].
- Poprawiona dokładność: Adaptacyjny charakter pozwala ADAPT-VQE odzyskać więcej energii korelacji w każdym kroku, co czyni go szczególnie skutecznym dla silnie skorelowanych układów, w których tradycyjny VQE ma trudności[8,9].
- Skalowalność i odporność na szum: Kompaktowy ansatz zmniejsza akumulację błędów bramek, zmniejsza narzut obliczeniowy i ogranicza liczbę parametrów wariacyjnych, które muszą być zminimalizowane.
ADAPT-VQE nadal nie jest doskonały. W niektórych przypadkach może zostać uwięziony lub spowolniony przez lokalne minima i może cierpieć z powodu nadmiernej parametryzacji. Może również być dość zasobochłonny, ponieważ wymaga obliczania gradientów i optymalizacji parametrów z wieloma strukturami bramek.
Estymacja fazy kwantowej (QPE)
QPE ma podobny cel jak VQE, ale bardzo różni się implementacją. QPE wymaga komputerów kwantowych odpornych na błędy ze względu na swoje na ogół głębokie obwody kwantowe i wysoki poziom koherencji, jakiego wymaga. Gdy QPE będzie można zaimplementować, będzie bardziej precyzyjne niż VQE. Jednym ze sposobów opisania różnicy jest precyzja jako funkcja głębokości obwodu. QPE osiąga precyzję przy głębokościach obwodu skalujących się jako [10]. VQE wymaga próbek, aby osiągnąć tę samą precyzję[10,11].
Krylov, SQD, QSCI i inne w tym kursie
VQE pomógł ustanowić algorytmy kwantowe, które nadal zależą od komputerów klasycznych, nie tylko do obsługi komputera kwantowego, ale także do znacznych części algorytmu. Kilka takich algorytmów jest przedmiotem pozostałej części tego kursu. Tutaj podajemy krótkie wyjaśnienie kilku z nich, po prostu aby porównać je z VQE. Zostaną one wyjaśnione znacznie bardziej szczegółowo w kolejnych lekcjach.
Kwantowa diagonalizacja Kryłowa (KQD)
Metody podprzestrzeni Krylov są sposobami rzutowania macierzy na podprzestrzeń w celu zmniejszenia jej wymiaru i uczynienia jej bardziej zarządzalną, przy jednoczesnym zachowaniu najważniejszych cech. Jednym z trików w tej metodzie jest wygenerowanie podprzestrzeni, która zachowuje te cechy; okazuje się, że generowanie tej podprzestrzeni jest ściśle związane z dobrze ugruntowaną metodą na komputerach kwantowych zwaną Trotteryzacją.
Istnieje kilka wariantów kwantowych metod Krylov, ale generalnie podejście jest następujące:
- Użyj komputera kwantowego do wygenerowania podprzestrzeni (podprzestrzeni Krylov) poprzez Trotteryzację
- Zrzutuj interesującą macierz na tę podprzestrzeń Krylov
- Diagonalizuj nowy zrzutowany Hamiltonian za pomocą komputera klasycznego
Kwantowa diagonalizacja oparta na próbkowaniu (SQD)
Kwantowa diagonalizacja oparta na próbkowaniu (SQD) jest związana z metodą Krylov w tym, że również próbuje zredukować wymiar macierzy do diagonalizacji, zachowując kluczowe cechy. SQD robi to w następujący sposób:
- Zacznij od początkowego przypuszczenia dla stanu podstawowego i przygotuj układ w tym stanie podstawowym.
- Użyj Sampler, aby pobrać ciągi bitów tworzące ten stan.
- Użyj zbioru stanów bazy obliczeniowej z samplera jako podprzestrzeni, na którą rzutujesz interesującą Cię macierz.
- Diagonalizuj mniejszą, zrzutowaną macierz za pomocą komputera klasycznego.
Jest to związane z VQE w tym, że wykorzystuje klasyczne i kwantowe obliczenia dla znacznych komponentów algorytmu. Oba mają również wspólny wymóg, abyśmy przygotowali dobre początkowe przypuszczenie lub ansatz. Ale rozkład pracy między komputerami klasycznymi i kwantowymi w SQD jest bardziej podobny do tego w metodzie Krylov.
W rzeczywistości metoda Krylov i SQD zostały niedawno połączone w metodę kwantowej diagonalizacji Krylov opartej na próbkowaniu (SKQD) [12].
Kwantowa interakcja konfiguracji podprzestrzeni
Kwantowa Wybrana Interakcja Konfiguracji (QSCI)[13] to algorytm, który wytwarza przybliżony stan podstawowy Hamiltonianu poprzez próbkowanie próbnej funkcji falowej w celu zidentyfikowania znaczących stanów bazy obliczeniowej do wygenerowania podprzestrzeni dla klasycznej diagonalizacji. Zarówno SQD, jak i QSCI używają komputera kwantowego do skonstruowania zredukowanej podprzestrzeni. Dodatkową siłą QSCI jest przygotowanie stanu, szczególnie w kontekście problemów chemicznych. Wykorzystuje różne strategie, takie jak stosowanie stanów ewoluowanych w czasie [14] oraz zestaw ansatzów inspirowanych chemią. Skupiając się na wydajnym przygotowaniu stanu, QSCI zmniejsza kwantowe koszty obliczeniowe dla Hamiltonianów chemicznych, zachowując jednocześnie wysoką wierność i wykorzystując odporność na szum z technik próbkowania stanów kwantowych [15]. QSCI zapewnia również adaptacyjną technikę konstrukcji, która zapewnia więcej ansatzów dla lepszego wyniku.
Domyślny przepływ pracy QSCI dla problemu chemicznego jest następujący:
- Zbuduj Hamiltonian molekularny za pomocą wybranego oprogramowania (takiego jak SciPy).
- Przygotuj algorytm QSCI, wybierając odpowiedni stan początkowy i ansatz inspirowany chemią z wstępnie wybranym zestawem parametrów.
- Próbkuj znaczące stany bazowe i diagonalizuj Hamiltonian za pomocą komputera klasycznego, aby uzyskać energię stanu podstawowego.
- Często stosuje się odzyskiwanie konfiguracji [16] i post-selekcję symetrii [15] jako technikę przetwarzania końcowego.
- Opcjonalnie przepływ pracy adaptacyjnego QSCI zawiera dodatkową pętlę optymalizacji od kroku 2 do kroku 3, przy użyciu większej liczby ansatzów z losowymi stanami początkowymi.
Sprawdź swoje zrozumienie
Przeczytaj poniższe pytania, przemyśl swoje odpowiedzi, a następnie kliknij trójkąty, aby odsłonić rozwiązania.
Co VQE ma wspólnego ze wszystkimi innymi wymienionymi powyżej metodami (z wyjątkiem QPE, który nie jest szczegółowo opisany)?
Odpowiedź:
Wszystkie wymagają pewnego rodzaju stanu próbnego lub funkcji falowej. Wszystkie działają najlepiej, gdy początkowe przypuszczenie dla tego stanu próbnego jest doskonałe.
Inną poprawną odpowiedzią jest to, że wszystkie są najłatwiejsze do zaimplementowania, gdy Hamiltonian jest łatwy do zmierzenia (można go posortować w stosunkowo niewiele grup komutujących operatorów Pauliego).
Co VQE ma wspólnego z żadną z innych wymienionych powyżej metod?
Odpowiedź:
Klasyczne optymalizatory. Żadna z pozostałych nie używa klasycznych algorytmów optymalizacyjnych do wyboru parametrów wariacyjnych.
Źródła
[2] https://en.wikipedia.org/wiki/Variational_quantum_eigensolver
[3] https://journals.aps.org/prapplied/abstract/10.1103/PhysRevApplied.19.024047
[4] https://arxiv.org/abs/2111.05176
[6] https://inquanto.quantinuum.com/tutorials/InQ_tut_fe4n2_2.html
[7] https://www.nature.com/articles/s41467-019-10988-2
[8] https://arxiv.org/abs/2210.15438
[9] https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.6.013254
[10] https://arxiv.org/html/2403.09624v1
[11] https://www.nature.com/articles/s42005-023-01312-y
[13] https://arxiv.org/abs/1802.00171
[14] https://arxiv.org/abs/2103.08505
[15] https://arxiv.org/html/2501.09702v1
[16] https://quri-sdk.qunasys.com/docs/examples/quri-algo-vm/qsci/
[17] https://arxiv.org/abs/2412.13839