Kwantowa diagonalizacja Kryłowa
W tej lekcji dotyczącej kwantowej diagonalizacji Krylov (KQD) odpowiemy na następujące pytania:
- Czym jest metoda Krylov w ogólnym ujęciu?
- Dlaczego metoda Krylov działa i pod jakimi warunkami?
- Jaką rolę odgrywa obliczanie kwantowe?
Część kwantowa obliczeń opiera się w dużej mierze na pracy z Ref [1].
Poniższe wideo przedstawia ogólny przegląd metod Krylov w obliczeniach klasycznych, motywuje ich zastosowanie oraz wyjaśnia, jaką rolę może odgrywać obliczanie kwantowe w tym nurcie pracy. W dalszym tekście znajdują się bardziej szczegółowe informacje oraz implementacja metody Krylov zarówno klasycznie, jak i przy użyciu komputera kwantowego.
1. Wprowadzenie do metod Krylov
Metoda podprzestrzeni Krylov może odnosić się do dowolnej z kilku metod zbudowanych wokół tak zwanej podprzestrzeni Krylov. Pełny przegląd tych metod wykracza poza zakres tej lekcji, lecz Ref [2-4] mogą dostarczyć znacznie szerszego kontekstu. Skupimy się tutaj na tym, czym jest podprzestrzeń Krylov, jak i dlaczego jest użyteczna w rozwiązywaniu problemów wartości własnych, a w końcu jak może być zaimplementowana na komputerze kwantowym. Definicja: Dla symetrycznej, dodatnio półokreślonej macierzy o wymiarze , przestrzeń Krylov rzędu jest przestrzenią rozpinaną przez wektory otrzymane przez mnożenie wyższych potęg macierzy , aż do , przez wektor odniesienia .
Chociaż wektory powyżej rozpinają to, co nazywamy podprzestrzenią Krylov, nie ma powodu sądzić, że będą one ortogonalne. Często stosuje się iteracyjny proces ortonormalizacji podobny do ortogonalizacji Grama-Schmidta. Tutaj proces jest nieco inny, ponieważ każdy nowy wektor jest czyniony ortogonalnym do pozostałych w momencie jego generowania. W tym kontekście nazywa się to iteracją Arnoldiego. Zaczynając od wektora początkowego , generuje się następny wektor , a następnie zapewnia się, że ten drugi wektor jest ortogonalny do pierwszego przez odjęcie jego rzutu na . Czyli
Łatwo teraz zauważyć, że ponieważ
Robimy to samo dla kolejnego wektora, zapewniając, że jest ortogonalny do obu poprzednich:
Jeśli powtórzymy ten proces dla wszystkich wektorów, otrzymamy kompletną ortonormalną bazę dla przestrzeni Krylov. Zauważ, że proces ortogonalizacji w tym miejscu daje zero, gdy , ponieważ ortogonalnych wektorów z konieczności rozpina pełną przestrzeń. Proces również da zero, jeśli dowolny wektor jest wektorem własnym , ponieważ wszystkie kolejne wektory będą wielokrotnościami tego wektora.
1.1 Prosty przykład: Krylov ręcznie
Przejdźmy krok po kroku przez generowanie podprzestrzeni Krylov na trywialnie małej macierzy, tak aby zobaczyć ten proces. Zaczynamy od interesującej nas początkowej macierzy :
Dla tego małego przykładu możemy łatwo wyznaczyć wektory i wartości własne nawet ręcznie. Tutaj pokazujemy rozwiązanie numeryczne.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy sympy
# One might use linalg.eigh here, but later matrices may not be Hermitian. So we use
# linalg.eig in this lesson.
import numpy as np
A = np.array([[4, -1, 0], [-1, 4, -1], [0, -1, 4]])
eigenvalues, eigenvectors = np.linalg.eig(A)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
The eigenvalues are [2.58578644 4. 5.41421356]
The eigenvectors are [[ 5.00000000e-01 -7.07106781e-01 5.00000000e-01]
[ 7.07106781e-01 1.37464400e-16 -7.07106781e-01]
[ 5.00000000e-01 7.07106781e-01 5.00000000e-01]]
Zapisujemy je tutaj do późniejszego porównania:
Chcielibyśmy zbadać, jak ten proces działa (lub zawodzi), w miarę jak zwiększamy wymiar naszej podprzestrzeni Krylov, . W tym celu zastosujemy następujący proces:
- Wygeneruj podprzestrzeń pełnej przestrzeni wektorowej, zaczynając od losowo wybranego wektora (nazwij go , jeśli jest już znormalizowany, jak powyżej).
- Zrzutuj pełną macierz na tę podprzestrzeń i znajdź wartości własne tej zrzutowanej macierzy .
- Zwiększ rozmiar podprzestrzeni, generując więcej wektorów, dbając o to, aby były one ortonormalne, przy użyciu procesu podobnego do ortogonalizacji Grama-Schmidta.
- Zrzutuj na większą podprzestrzeń i znajdź wartości własne otrzymanej macierzy .
- Powtarzaj to, aż wartości własne zbiegną się (lub, w tym zabawkowym przypadku, aż wygenerujesz wektory rozpinające pełną przestrzeń wektorową oryginalnej macierzy ).
Normalna implementacja metody Krylov nie musiałaby rozwiązywać problemu wartości własnych dla macierzy rzutowanej na każdą podprzestrzeń Krylov w miarę jej budowania. Można by skonstruować podprzestrzeń o żądanym wymiarze, zrzutować macierz na tę podprzestrzeń i zdiagonalizować zrzutowaną macierz. Rzutowanie i diagonalizowanie przy każdym wymiarze podprzestrzeni jest wykonywane tylko w celu sprawdzenia zbieżności.
Wymiar :
Wybieramy losowy wektor, powiedzmy
Jeśli nie jest on już znormalizowany, znormalizuj go.
Teraz rzutujemy naszą macierz na podprzestrzeń tego jednego wektora:
To jest nasze rzutowanie macierzy na naszą podprzestrzeń Krylov, gdy zawiera ona tylko pojedynczy wektor, . Wartość własna tej macierzy wynosi trywialnie 4. Możemy traktować to jako nasze oszacowanie zerowego rzędu wartości własnych (w tym przypadku tylko jednej) macierzy . Chociaż jest to słabe oszacowanie, jest to poprawny rząd wielkości.
Wymiar :
Teraz generujemy kolejny wektor w naszej podprzestrzeni poprzez działanie na poprzedni wektor:
Teraz odejmujemy rzut tego wektora na poprzedni wektor, aby zapewnić ortogonalność.
Jeśli nie jest już znormalizowany, znormalizuj go. W tym przypadku wektor był już znormalizowany, więc
Teraz rzutujemy naszą macierz A na podprzestrzeń tych dwóch wektorów:
Pozostaje nam jeszcze problem wyznaczenia wartości własnych tej macierzy. Ale ta macierz jest nieco mniejsza niż pełna macierz. W problemach dotyczących bardzo dużych macierzy praca z tą mniejszą podprzestrzenią może być niezwykle korzystna.
Chociaż nadal nie jest to dobre oszacowanie, jest lepsze niż oszacowanie zerowego rzędu. Wykonamy jeszcze jedną iterację, aby upewnić się, że proces jest jasny. Jednak podważa to sens metody, ponieważ w następnej iteracji będziemy diagonalizować macierz 3x3, co oznacza, że nie zaoszczędziliśmy czasu ani mocy obliczeniowej.
Wymiar :
Teraz generujemy kolejny wektor w naszej podprzestrzeni poprzez działanie A na poprzedni wektor:
Teraz odejmujemy rzut tego wektora na oba nasze poprzednie wektory, aby zapewnić ortogonalność.
Jeśli nie jest już znormalizowany, znormalizuj go. W tym przypadku wektor był już znormalizowany, więc
Teraz rzutujemy naszą macierz na podprzestrzeń tych wektorów:
Wyznaczamy teraz wartości własne:
Te wartości własne są dokładnie wartościami własnymi macierzy oryginalnej . Tak musi być, ponieważ rozszerzyliśmy naszą podprzestrzeń Krylov, by rozpinała całą przestrzeń wektorową macierzy oryginalnej .
W tym przykładzie metoda Krylov może nie wydawać się szczególnie łatwiejsza niż bezpośrednia diagonalizacja. W istocie, jak zobaczymy w kolejnych sekcjach, metoda Krylov jest korzystna dopiero powyżej pewnego wymiaru macierzy; ma ona nam pomóc w rozwiązywaniu problemów wartości własnych/wektorów własnych niezwykle dużych macierzy.
To jedyny przykład, który pokażemy rozwiązany „ręcznie”, ale sekcja 2 poniżej przedstawia przykłady obliczeniowe.
Wyjaśnienie terminów
Powszechnym nieporozumieniem jest przekonanie, że dla danego problemu istnieje tylko jedna podprzestrzeń Krylov. Jednak oczywiście, ponieważ istnieje wiele wektorów początkowych, do których można zastosować naszą macierz, istnieje wiele możliwych podprzestrzeni Krylov. Wyrażenia „the Krylov subspace” („ta podprzestrzeń Krylov”) będziemy używać wyłącznie w odniesieniu do konkretnej podprzestrzeni Krylov już zdefiniowanej dla konkretnego przykładu. Dla ogólnych podejść do rozwiązywania problemów będziemy mówić o „a Krylov subspace” („pewnej podprzestrzeni Krylov”). Ostatnie wyjaśnienie: poprawne jest także odwoływanie się do „przestrzeni Krylov”. Często spotyka się określenie „podprzestrzeń Krylov” ze względu na jej zastosowanie w kontekście rzutowania macierzy z przestrzeni początkowej na podprzestrzeń. Zgodnie z tym kontekstem będziemy tu najczęściej mówić o podprzestrzeni.
Sprawdź swoje zrozumienie
Przeczytaj poniższe pytania, zastanów się nad odpowiedzią, a następnie kliknij trójkąt, aby odsłonić każde rozwiązanie.
Wyjaśnij, dlaczego (a) nie jest użyteczne i (b) nie jest możliwe rozszerzanie wymiaru podprzestrzeni Krylov ponad wymiar rozpatrywanej macierzy.
Odpowiedź:
(a) Ponieważ ortonormalizujemy wektory w miarę ich wytwarzania, zbiór takich wektorów utworzy bazę kompletną, co oznacza, że ich liniowa kombinacja może zostać użyta do utworzenia dowolnego wektora w przestrzeni. (b) Proces ortogonalizacji polega na odejmowaniu rzutu nowego wektora na wszystkie poprzednie wektory. Jeżeli wszystkie poprzednie wektory rozpinają pełną przestrzeń wektorową, to odejmowanie rzutów na pełną podprzestrzeń zawsze pozostawi nam wektor zerowy.
Przypuśćmy, że kolega-badacz demonstruje metodę Krylov zastosowaną do niewielkiej macierzy przykładowej kilku współpracownikom. Ów kolega-badacz planuje użyć macierzy i wektora początkowego:
oraz
Czy coś jest z tym nie tak? Jak doradziłbyś swojemu koledze?
Odpowiedź:
Twój kolega przypadkowo wybrał wektor własny jako swój wektor początkowy. Działanie macierzą na wektor początkowy po prostu zwróci ten sam wektor, przeskalowany o wartość własną. Nie wygeneruje to podprzestrzeni o rosnącym wymiarze. Doradź koledze wybór innego wektora początkowego, upewniając się, że nie jest on wektorem własnym.
Zastosuj metodę Krylov do poniższej macierzy, wybierając odpowiedni nowy wektor początkowy. Zapisz oszacowania minimalnej wartości własnej w 0-tym i 1-szym rzędzie Twojej podprzestrzeni Krylov.
Odpowiedź:
Istnieje wiele możliwych odpowiedzi w zależności od wyboru wektora początkowego. Wybierzemy:
Aby otrzymać , stosujemy jednokrotnie do , a następnie czynimy ortogonalnym do
W 0-tym rzędzie rzut na naszą podprzestrzeń Krylov wynosi
W 1-szym rzędzie rzut na tę podprzestrzeń Krylov wynosi
Można to wykonać ręcznie, ale najłatwiej zrobić to przy użyciu numpy:
import numpy as np
vstar = np.array([[1/np.sqrt(3),1/np.sqrt(3),1/np.sqrt(3)],[-1/np.sqrt(6),np.sqrt(2/3),-1/np.sqrt(6)]]
)
A = np.array([[1, 1, 0],
[1, 1, 1],
[0, 1, 1]])
v = np.array([[1/np.sqrt(3),-1/np.sqrt(6)],[1/np.sqrt(3),np.sqrt(2/3)],[1/np.sqrt(3),-1/np.sqrt(6)]])
proj = vstar@A@v
print(proj)
eigenvalues, eigenvectors = np.linalg.eig(proj)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
zwraca:
[[ 2.33333333 0.47140452]
[ 0.47140452 -0.33333333]]
The eigenvalues are [ 2.41421356 -0.41421356]
The eigenvectors are [[ 0.98559856 -0.16910198]
[ 0.16910198 0.98559856]]
Oszacowanie minimalnej wartości własnej wynosi -0.414.
1.2 Rodzaje metod Krylov
„Metody podprzestrzeni Krylov” mogą odnosić się do dowolnej z kilku technik iteracyjnych stosowanych do rozwiązywania dużych układów liniowych i problemów wartości własnych. Wspólną ich cechą jest to, że konstruują przybliżone rozwiązanie z podprzestrzeni Krylov
gdzie to przybliżenie początkowe (zob. odn. [5]). Różnią się one sposobem wyboru najlepszego przybliżenia z tej podprzestrzeni, równoważąc takie czynniki jak szybkość zbieżności, zużycie pamięci i ogólny koszt obliczeniowy. Celem tej lekcji jest wykorzystanie obliczeń kwantowych w kontekście metod podprzestrzeni Krylov; wyczerpujące omówienie tych metod wykracza poza jej zakres. Poniższe krótkie definicje mają charakter wyłącznie kontekstowy i zawierają pewne odnośniki do dalszego zgłębiania tych metod.
Metoda gradientów sprzężonych (CG): Metoda ta jest używana do rozwiązywania symetrycznych, dodatnio określonych układów liniowych[6]. Minimalizuje ona normę A błędu w każdej iteracji, co czyni ją szczególnie skuteczną dla układów powstających z dyskretyzacji eliptycznych równań różniczkowych cząstkowych[7]. Użyjemy tego podejścia w następnej sekcji, aby uzasadnić, dlaczego podprzestrzeń Krylov byłaby efektywną podprzestrzenią, w której można poszukiwać lepszych rozwiązań układów liniowych.
Metoda uogólnionej minimalnej reszty (GMRES): Została zaprojektowana do rozwiązywania ogólnych niesymetrycznych układów liniowych. Minimalizuje normę reszty nad przestrzenią Krylov w każdej iteracji, co czyni ją solidną, lecz potencjalnie pamięciochłonną dla dużych układów[7].
Metoda minimalnej reszty (MINRES): Metoda ta jest używana do rozwiązywania symetrycznych nieokreślonych układów liniowych. Jest podobna do GMRES, ale wykorzystuje symetrię macierzy w celu zmniejszenia kosztu obliczeniowego[8].
Inne godne uwagi podejścia to metoda pełnej ortogonalizacji (FOM), ściśle powiązana z metodą Arnoldiego dla problemów wartości własnych, metoda bi-sprzężonych gradientów (BiCG) oraz metoda indukowanej redukcji wymiaru (IDR).
1.3 Dlaczego metoda podprzestrzeni Krylov działa
Tutaj uzasadnimy, że metoda podprzestrzeni Krylov powinna być efektywnym sposobem przybliżania wartości własnych macierzy poprzez iteracyjne udoskonalanie przybliżeń wektorów własnych, z perspektywy najszybszego spadku. Będziemy argumentować, że przy zadanym początkowym przybliżeniu stanu podstawowego, przestrzenią kolejnych poprawek do tego początkowego przybliżenia zapewniającą najszybszą zbieżność jest podprzestrzeń Krylov. Nie posuwamy się do rygorystycznego dowodu zachowania zbieżności.
Załóżmy, że nasza rozpatrywana macierz jest symetryczna i dodatnio określona. Czyni to nasze rozumowanie najbardziej adekwatnym dla powyższej metody CG. Nie czynimy tu żadnych założeń dotyczących rzadkości; nie twierdzimy również, że musi być macierzą hermitowską (co jest wymagane, jeśli ma ona być Hamiltonianem).
Zazwyczaj chcemy rozwiązać problem postaci
Można by sobie wyobrazić, że , gdzie to pewna stała, jak w problemie wartości własnych. Nasze sformułowanie problemu pozostaje jednak na razie bardziej ogólne.
Zaczynamy od wektora , który jest przybliżonym rozwiązaniem. Mimo że istnieją analogie między tym przybliżeniem a z Sekcji 1.1, nie wykorzystujemy ich tutaj. Nasze przybliżenie obarczone jest błędem, który nazwiemy
Definiujemy również resztę
Używamy tutaj dużej litery , aby odróżnić resztę od wymiaru naszej podprzestrzeni Krylov .

Chcemy teraz wykonać krok korekcyjny postaci
o którym mamy nadzieję, że poprawi nasze przybliżenie. Tutaj jest pewnym wektorem jeszcze do wyznaczenia. Niech będzie błędem po dokonaniu korekty. Wówczas

Interesuje nas, jak zachowuje się nasz błąd przekształcony przez naszą macierz. Obliczmy zatem normę błędu. To jest
gdzie wykorzystaliśmy symetrię oraz fakt, że Tutaj to pewna stała niezależna od . Jak wspomniano w Sekcji 1.2, norma błędu nie jest jedyną wielkością, jaką moglibyśmy wybrać do minimalizacji, ale jest dobrą miarą. Chcemy zobaczyć, jak ta wielkość zmienia się wraz z naszym wyborem wektorów korekcyjnych Definiujemy zatem funkcję poprzez ustalenie
jest po prostu błędem jako funkcją poprawki mierzoną w normie . Chcemy zatem wybrać tak, aby było możliwie jak najmniejsze. W tym celu obliczamy gradient . Korzystając z symetrii mamy
Gradient wskazuje kierunek najszybszego wzrostu, co oznacza, że jego przeciwieństwo daje nam kierunek, w którym funkcja maleje najszybciej: kierunek najszybszego spadku. W naszym początkowym przybliżeniu , gdzie , mamy Zatem funkcja maleje najszybciej w kierunku residuum Nasz początkowy wybór najbardziej skorzystałby więc z dodania wektora dla pewnego skalaru .
W następnym kroku ponownie wybieramy wektor i dodajemy jego wartość do bieżącego przybliżenia. Używając tego samego argumentu co poprzednio, wybieramy dla pewnego skalaru . Kontynuujemy w ten sposób, tak że iteracja naszego wektora ma postać
Równoważnie, chcemy zbudować przestrzeń, z której wybieramy nasze ulepszone oszacowania, poprzez dodawanie kolejno , i tak dalej. oszacowany wektor leży w
Teraz, korzystając z relacji
widzimy, że
Oznacza to, że przestrzeń, którą budujemy, najefektywniej przybliżająca poprawne rozwiązanie , jest dokładnie przestrzenią zbudowaną przez kolejne działania macierzy na Podprzestrzeń Krylov jest przestrzenią rozpinaną przez wektory kolejnych kierunków najszybszego spadku.
Na koniec powtarzamy, że nie sformułowaliśmy żadnych twierdzeń liczbowych dotyczących skalowania tego podejścia, ani nie omówiliśmy porównawczej korzyści dla macierzy rzadkich. Ma to jedynie uzasadnić użycie metod podprzestrzeni Krylov i dostarczyć pewnej intuicji na ich temat. Teraz zbadamy numerycznie zachowanie tych metod.
Sprawdź swoje zrozumienie
Przeczytaj poniższe pytanie, zastanów się nad odpowiedzią, a następnie kliknij trójkąt, aby ujawnić rozwiązanie.
W przedstawionym powyżej przepływie pracy zaproponowaliśmy minimalizację normy błędu. Jakie inne wielkości można by rozważyć minimalizację przy poszukiwaniu stanu podstawowego i jego wartości własnej?
Odpowiedź:
Można sobie wyobrazić użycie wektora residualnego zamiast normy błędu. Mogą istnieć przypadki, w których rozważenie samego wektora błędu jest przydatne.
2. Metody Krylov w obliczeniach klasycznych
W tej sekcji implementujemy obliczeniowo iteracje Arnoldiego, abyśmy mogli wykorzystać podprzestrzeń Krylov do rozwiązywania zagadnień własnych. Zastosujemy to najpierw do niewielkiego przykładu, a następnie zbadamy, jak czas obliczeń skaluje się wraz ze wzrostem rozmiaru rozważanej macierzy. Kluczową ideą będzie tutaj to, że generowanie wektorów rozpinających przestrzeń Krylov będzie w znacznym stopniu wpływać na całkowity wymagany czas obliczeń. Wymagana pamięć będzie się różnić między poszczególnymi metodami Krylov. Jednak ograniczenia pamięci mogą ograniczać zastosowanie tradycyjnych metod Krylov.
2.1 Prosty przykład małej skali
W procesie tworzenia podprzestrzeni Krylov będziemy musieli ortonormalizować wektory w naszej podprzestrzeni. Zdefiniujmy funkcję, która przyjmuje ustalony wektor z naszej podprzestrzeni vknown (nie zakładamy, że jest znormalizowany) oraz kandydata na wektor do dodania do naszej podprzestrzeni vnext i sprawia, że vnext jest ortogonalny do vknown oraz znormalizowany. Zdefiniujmy dalej funkcję, która przechodzi przez ten proces dla wszystkich ustalonych wektorów w naszej podprzestrzeni Krylov, aby zapewnić w pełni ortonormalny zbiór.
# vknown is some established vector in our subspace. vnext is one we wish to add,
# which must be orthogonal to vknown.
def orthog_pair(vknown, vnext):
vknown = vknown / np.sqrt(vknown.T @ vknown)
diffvec = vknown.T @ vnext * vknown
vnext = vnext - diffvec
return vnext
# v is the candidate vector to be added to our subspace. s is the existing subspace.
def orthoset(v, s):
v = v / np.sqrt(v.T @ v)
temp = v
for i in range(len(s)):
temp = orthog_pair(s[i], temp)
v = temp / np.sqrt(temp.T @ temp)
return v
Teraz zdefiniujmy funkcję, która iteracyjnie buduje coraz większą podprzestrzeń Krylov, aż przestrzeń wektorów Krylov będzie rozpinać całą przestrzeń pierwotnej macierzy. Umożliwi nam to sprawdzenie, jak dobrze wartości własne otrzymane przy użyciu naszej metody podprzestrzeni Krylov pasują do wartości dokładnych w funkcji wymiaru podprzestrzeni Krylov. Co istotne, nasza funkcja krylov_full_build zwraca wektory Krylov, rzutowane Hamiltoniany, wartości własne oraz wymagany czas.
# Necessary imports and definitions to track time in microseconds
import time
def time_mus():
return int(time.time() * 1000000)
# This function constructs a Krylov subspace that spans the whole space of the original matrix.
# Input:
# v0 : initial vector
# matrix : original matrix to be diagonalized
# Output:
# ks : Krylov vectors
# Hs : projected Hamiltonians
# eigs : eigenvalues
# k_tot_times : time required for the operation
def krylov_full_build(v0, matrix):
t0 = time_mus()
b = v0 / np.sqrt(v0 @ v0.T)
A = matrix
ks = []
ks.append(b)
Hs = []
eigs = []
Hs.append(b.T @ A @ b)
eigs.append(np.array([b.T @ A @ b]))
k_tot_times = []
for j in range(len(A) - 1):
vec = A @ ks[j].T
ortho = orthoset(vec, ks)
ks.append(ortho)
ksarray = np.array(ks)
Hs.append(ksarray @ A @ ksarray.T)
eigs.append(np.linalg.eig(Hs[j + 1]).eigenvalues)
k_tot_times.append(time_mus() - t0)
# Return the Krylov vectors, the projected Hamiltonians, the eigenvalues,
# and the total time required.
return (ks, Hs, eigs, k_tot_times)
Przetestujemy to na macierzy, która jest wciąż dość mała, ale większa niż taka, którą chcielibyśmy rozwiązywać ręcznie.
# Define our small test matrix
test_matrix = np.array(
[
[4, -1, 0, 1, 0],
[-1, 4, -1, 2, 1],
[0, -1, 4, 3, 3],
[1, 2, 3, 4, 0],
[0, 1, 3, 0, 4],
]
)
# Give the test matrix and an initial guess as arguments in the function defined above.
# Calculate outputs.
test_ks, test_Hs, test_eigs, text_k_tot_times = krylov_full_build(
np.array([0.5, 0.5, 0, 0.5, 0.5]), test_matrix
)
Nasze funkcje możemy sprawdzić, upewniając się, że w ostatnim kroku (gdy przestrzeń Krylov jest pełną przestrzenią wektorową pierwotnej macierzy) wartości własne z metody Krylov dokładnie odpowiadają tym z dokładnej diagonalizacji numerycznej:
print(np.linalg.eig(test_matrix).eigenvalues)
print(test_eigs[len(test_matrix) - 1])
[-1.36956923 8.43756009 2.9040308 5.34436028 4.68361806]
[-1.36956923 8.43756009 2.9040308 4.68361806 5.34436028]
To się udało. Oczywiście, to, co naprawdę ma znaczenie, to jak dobre jest nasze przybliżenie w funkcji wymiaru naszej podprzestrzeni Krylov. Ponieważ często interesuje nas znajdowanie stanów podstawowych i innych minimalnych wartości własnych (a także z innych, bardziej algebraicznych powodów wyjaśnionych poniżej), przyjrzyjmy się naszemu oszacowaniu najmniejszej wartości własnej w funkcji wymiaru podprzestrzeni Krylov. Mianowicie:
def errors(matrix, krylov_eigs):
targ_min = min(np.linalg.eig(matrix).eigenvalues)
err = []
for i in range(len(matrix)):
err.append(min(krylov_eigs[i]) - targ_min)
return err
import matplotlib.pyplot as plt
krylov_error = errors(test_matrix, test_eigs)
plt.plot(krylov_error)
plt.axhline(y=0, color="red", linestyle="--") # Add dashed red line at y=0
plt.xlabel("Order of Krylov subspace") # Add x-axis label
plt.ylabel("Error in minimum eigenvalue") # Add y-axis label
plt.show()
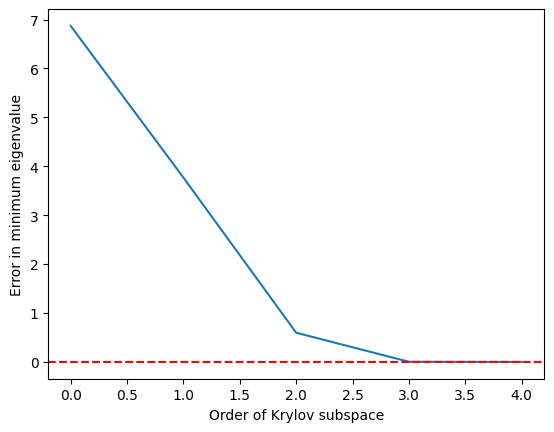
Widzimy, że minimalna wartość własna jest osiągana dość dokładnie, gdy podprzestrzeń Krylov urośnie do a jest idealna przy
2.2 Skalowanie czasu z wymiarem macierzy
Przekonajmy się, że metoda Krylov może być korzystna w porównaniu z dokładnymi numerycznymi solverami wartości własnych, w następujący sposób:
- Konstruujemy losowe macierze (nierzadkie, nie idealne zastosowanie dla KQD)
- Wyznaczamy wartości własne za pomocą dwóch metod: bezpośrednio używając NumPy oraz wykorzystując podprzestrzeń Krylov.
- Wybieramy próg określający, jak dokładne muszą być nasze wartości własne, zanim zaakceptujemy oszacowania Krylov.
- Porównujemy czas rzeczywisty potrzebny do rozwiązania tymi dwoma sposobami.
Zastrzeżenia: Jak omówimy szczegółowo poniżej, kwantowa diagonalizacja Krylov najlepiej stosuje się do operatorów, których reprezentacje macierzowe są rzadkie i/lub mogą być zapisane przy użyciu niewielkiej liczby grup komutujących operatorów Pauli. Losowe macierze, których tu używamy, nie pasują do tego opisu. Są one użyteczne jedynie do zbadania skali, w której klasyczne metody Krylov mogłyby być przydatne. Po drugie, używając metody Krylov, będziemy obliczać wartości własne przy użyciu wielu podprzestrzeni Krylov o różnych rozmiarach. Podamy czas wymagany dla podprzestrzeni Krylov o minimalnym wymiarze, która osiąga naszą wymaganą dokładność dla wartości własnej stanu podstawowego. Ponownie, różni się to nieco od rozwiązywania problemu niemożliwego do rozwiązania przez dokładne solvery wartości własnych, ponieważ używamy dokładnego rozwiązania do oceny potrzebnego wymiaru.
Zaczynamy od wygenerowania naszego zbioru macierzy losowych.
import numpy as np
# Set the random seed
np.random.seed(42)
# how many random matrices will we make
num_matrix = 200
matrices = []
for m in range(1, num_matrix):
matrices.append(np.random.rand(m, m))
Teraz diagonalizujemy każdą macierz bezpośrednio, używając numpy. Obliczamy czas potrzebny do diagonalizacji w celu późniejszego porównania.
matrix_numpy_times = []
matrix_numpy_eigs = []
for mm in range(num_matrix - 1):
t0 = time_mus()
matrix_numpy_eigs.append(min(np.linalg.eig(matrices[mm]).eigenvalues))
matrix_numpy_times.append(time_mus() - t0)
plt.plot(matrix_numpy_times)
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()
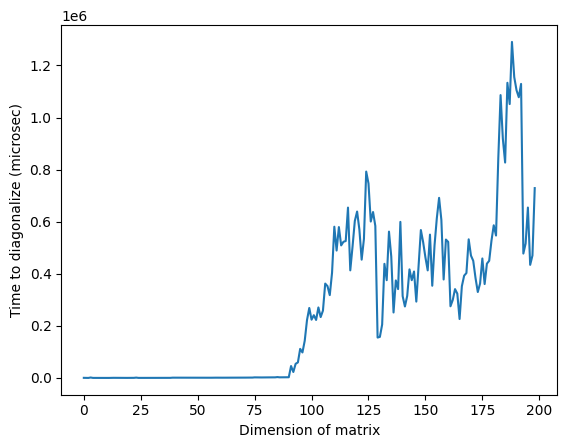
Zauważ, że na powyższym obrazie anomalnie wysoki czas w okolicach wymiaru 125 może wynikać z losowej natury macierzy lub z implementacji na użytym klasycznym procesorze, ale nie jest on odtwarzalny. Ponowne uruchomienie kodu da inny profil z innymi anomalnymi pikami.
Teraz dla każdej macierzy zbudujemy podprzestrzeń Krylova i będziemy krokowo obliczać wartości własne. W każdym kroku sprawdzimy, czy najniższa wartość własna została uzyskana z zadaną bezwzględną dokładnością. Podprzestrzeń, która po raz pierwszy daje nam wartości własne w zadanym zakresie błędu, jest podprzestrzenią, dla której zapiszemy czasy obliczeń. Wykonanie tej komórki może zająć kilka minut, w zależności od szybkości procesora. Możesz pominąć wykonanie lub zmniejszyć maksymalny wymiar diagonalizowanych macierzy. Spojrzenie na wstępnie obliczone wyniki jest wystarczające.
# Choose the absolute error you can tolerate, and make a list for tracking the Krylov subspace size
# at which that error is achieved.
abserr = 0.05
accept_subspace_size = []
# Lists to store total time spent on the Krylov method, and the subset of that time spent on
# diagonalizing the projected matrix.
matrix_krylov_tot_times = []
matrix_krylov_dim = []
# Step through all our random matrices
for mm in range(0, num_matrix - 1):
test_ks, test_Hs, test_eigs, test_k_tot_times = krylov_full_build(
np.ones(len(matrices[mm])), matrices[mm]
)
# We have not yet found a Krylov subspace that produces our minimum eigenvalue to
# within the required error.
found = 0
for j in range(0, len(matrices[mm]) - 1):
# If we still haven't found the desired subspace...
if found == 0:
# ...but if this one satisfies the requirement, then record everything
if (
abs((min(test_eigs[j]) - matrix_numpy_eigs[mm]) / matrix_numpy_eigs[mm])
< abserr
):
accept_subspace_size.append(j)
matrix_krylov_tot_times.append(test_k_tot_times[j])
matrix_krylov_dim.append(mm)
found = 1
Wykreślmy czasy uzyskane dla tych dwóch metod w celu porównania:
plt.plot(matrix_numpy_times, color="blue")
plt.plot(matrix_krylov_dim, matrix_krylov_tot_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()

Są to rzeczywiste wymagane czasy, ale dla potrzeb dyskusji wygładźmy te krzywe, uśredniając po kilku sąsiednich punktach / wymiarach macierzy. Poniżej pokazano, jak to zrobić:
smooth_numpy_times = []
smooth_krylov_times = []
# Choose the number of adjacent points over which to average forward;
# the same will be used backward.
smooth_steps = 10
# We will do this smoothing for all points/matrix dimensions
for i in range(len(matrix_krylov_tot_times)):
# Ensure we don't exceed the boundaries of our lists
start = max(0, i - smooth_steps)
end = min(len(matrix_krylov_tot_times) - 1, i + smooth_steps)
# Dummy variables for accumulating an average over adjacent points. This is done for both Krylov
# and the NumPy calculations.
smooth_count = 0
smooth_numpy_sum = 0
smooth_krylov_sum = 0
for j in range(start, end):
smooth_numpy_sum = smooth_numpy_sum + matrix_numpy_times[j]
smooth_krylov_sum = smooth_krylov_sum + matrix_krylov_tot_times[j]
smooth_count = smooth_count + 1
# Appending the averaged adjacent values to our new smooth lists
smooth_numpy_times.append(smooth_numpy_sum / smooth_count)
smooth_krylov_times.append(smooth_krylov_sum / smooth_count)
plt.plot(smooth_numpy_times, color="blue")
plt.plot(smooth_krylov_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (smoothed, microsec)") # Add y-axis label
plt.show()

Zauważ, że czas potrzebny na zbudowanie podprzestrzeni Krylova początkowo przewyższa czas potrzebny na pełną diagonalizację w numpy. Jednak wraz ze wzrostem rozmiaru macierzy metoda Krylova staje się korzystniejsza. Jest to prawdą nawet wtedy, gdy obniżymy nasz dopuszczalny błąd, ale przewaga pojawia się przy większym rozmiarze macierzy. Warto to przeanalizować.
Złożoność czasowa numerycznej diagonalizacji wynosi (z pewnymi różnicami między algorytmami). Złożoność czasowa generowania bazy ortonormalnej złożonej z wektorów również wynosi . A więc przewaga metody Krylova nie jest związana z użyciem bazy ortonormalnej, lecz z użyciem konkretnej bazy ortonormalnej, która skutecznie wyodrębnia interesujące nas wartości własne. Widzieliśmy to już w szkicu dowodu w pierwszej sekcji tej lekcji, i jest to krytyczne dla gwarancji zbieżności w metodach Krylova. Podsumujmy nasze dotychczasowe postępy:
- Dla bardzo dużych macierzy metoda podprzestrzeni Krylova może dawać przybliżone wartości własne z wymaganą tolerancją szybciej niż tradycyjne algorytmy diagonalizacji.
- Dla takich bardzo dużych macierzy generowanie podprzestrzeni Krylova jest najbardziej czasochłonną częścią metody podprzestrzeni Krylova.
- Dlatego wydajny sposób generowania podprzestrzeni Krylova byłby bardzo cenny. W tym miejscu do gry wkracza wreszcie komputer kwantowy.
Sprawdź swoje zrozumienie
Przeczytaj poniższe pytanie, zastanów się nad odpowiedzią, a następnie kliknij trójkąt, aby odkryć rozwiązanie.
Odwołaj się do wygładzonego wykresu czasów diagonalizacji w zależności od wymiaru macierzy powyżej.
(a) Przy jakim w przybliżeniu wymiarze macierzy metoda Krylova stała się szybsza według tego wykresu?
(b) Jakie aspekty obliczeń mogłyby zmienić wymiar, przy którym metoda Krylova staje się szybsza?
Odpowiedź:
(a) Odpowiedzi mogą się różnić, jeśli ponownie uruchomisz obliczenia, ale metoda Krylova staje się szybsza w przybliżeniu przy wymiarze 80-85. (b) Istnieje wiele możliwych odpowiedzi. Ważnymi czynnikami są wymagana precyzja oraz rzadkość diagonalizowanych macierzy.
3. Krylov poprzez ewolucję w czasie
Wszystko, co dotychczas opisaliśmy, można zrobić klasycznie. Jak więc i kiedy użylibyśmy komputera kwantowego? Dla bardzo dużych macierzy metoda Krylova może wymagać długich czasów obliczeń i dużej ilości pamięci. Czas potrzebny na operację macierzową na skaluje się jak w najgorszym przypadku. Nawet mnożenie rzadkich macierzy przez wektor (typowy przypadek w klasycznych rozwiązywaczach typu Krylova) ma złożoność czasową skalującą się jak . Jest to wykonywane dla każdego wektora, który chcemy mieć w naszej podprzestrzeni. Wymiar podprzestrzeni zazwyczaj nie stanowi znaczącej frakcji i często skaluje się jak . Zatem generowanie wszystkich wektorów skaluje się jak w najgorszym przypadku. Chociaż są inne kroki, takie jak ortogonalizacja, to jest dominujące skalowanie, które należy mieć na uwadze.
Obliczenia kwantowe pozwalają nam zmienić, jakie atrybuty problemu determinują skalowanie czasu i wymaganych zasobów. Zamiast zależności od rozmiaru macierzy na każdym kroku, zobaczymy takie rzeczy jak liczba pomiarów (shotów) i liczba niekomutujących członów Pauliego, które tworzą Hamiltonian. Przyjrzyjmy się, jak to działa.
3.1 Ewolucja w czasie
Przypomnijmy, że operator ewolucji stanu kwantowego w czasie to (i bardzo powszechne, zwłaszcza w obliczeniach kwantowych, jest pomijanie w notacji). Jednym ze sposobów zrozumienia, a nawet realizacji takiej funkcji wykładniczej operatora jest spojrzenie na jej rozwinięcie w szereg Taylora. Zauważmy, że ta operacja działająca na pewnym początkowym wektorze daje sumę wyrazów z rosnącymi potęgami zastosowanymi do stanu początkowego. Wygląda na to, że możemy po prostu stworzyć naszą podprzestrzeń Krylova, ewoluując w czasie nasz początkowy stan próbny!
Zastrzeżenie polega na realizacji ewolucji w czasie na prawdziwym komputerze kwantowym. Wiele członów w Hamiltonianie nie będzie ze sobą komutować. A więc, podczas gdy niektóre proste operatory wykładnicze, takie jak , odpowiadają prostym obwodom, ogólne Hamiltoniany już nie. A ponieważ zawierają niekomutujące człony, nie możemy po prostu rozłożyć funkcji wykładniczej na iloczyn prostych, tak jak to robimy z liczbami.
Nie jest to więc trywialne, ale jest to dobrze zbadany proces w obliczeniach kwantowych. Ewolucję w czasie na komputerach kwantowych realizujemy za pomocą procesu zwanego trotteryzacją, która sama w sobie jest bogatym tematem[10]. Ale na bardzo wysokim poziomie, dzieląc ewolucję w czasie na bardzo małe kroki, powiedzmy kroków o rozmiarze , ograniczamy skutki niekomutatywności członów.
gdzie .
Nazwijmy podprzestrzeń Krylova rzędu r, którą wygenerowaliśmy w kontekście klasycznym, bezpośrednio używając potęg H, „potęgową podprzestrzenią Krylova”.
Teraz wygenerujemy podobną przestrzeń, używając unitarnego operatora ewolucji czasowej ; będziemy się do niej odnosić jako do „unitarnej przestrzeni Krylova” . Potęgowa podprzestrzeń Krylova , której używamy klasycznie, nie może być wygenerowana bezpośrednio na komputerze kwantowym, ponieważ nie jest operatorem unitarnym. Można wykazać, że użycie unitarnej podprzestrzeni Krylova daje podobne gwarancje zbieżności jak potęgowa podprzestrzeń Krylova, mianowicie błąd stanu podstawowego zbiega się efektywnie, o ile stan początkowy ma z prawdziwym stanem podstawowym nakrycie, które nie zanika wykładniczo, i o ile istnieje wystarczająca przerwa między wartościami własnymi. Zobacz Odnośnik [1], aby zapoznać się z dokładniejszą dyskusją na temat zbieżności.
Tutaj potęgi stają się różnymi krokami czasowymi (-ta potęga to postęp o czas ). Element podprzestrzeni, który podlega ewolucji czasowej przez łączny czas , możemy oznaczyć jako .
Możemy rzutować nasz Hamiltonian H na unitarną podprzestrzeń Krylova, . Innymi słowy, obliczamy każdy element macierzy w bazie . Tę rzutowaną macierz będziemy oznaczać jako .
3.2 Jak zaimplementować na komputerze kwantowym
Elementy macierzy są dane przez wartości oczekiwane , które można oszacować za pomocą komputera kwantowego. Należy pamiętać, że można zapisać jako sumę operatorów Pauli działających na różne kubity, oraz że nie wszystkie operatory Pauli można mierzyć jednocześnie. Możemy posortować wyrazy Pauli w grupy wyrazów komutujących i mierzyć je wszystkie naraz. Może nam jednak być potrzebnych wiele takich grup, aby objąć wszystkie wyrazy. Tak więc istotna staje się liczba rozłącznych grup komutujących, na które można rozbić wyrazy, .
Tutaj jest ciągiem Pauli postaci lub zbiorem takich ciągów Pauli, które wzajemnie komutują. Zakładając, że możemy zapisać jako taką sumę mierzalnych operatorów, następujące wyrażenia na elementy macierzowe mogą być zrealizowane za pomocą prymitywu Estimator z Qiskit Runtime.
Gdzie są wektorami unitarnej przestrzeni Krylova, a są wielokrotnościami wybranego kroku czasowego . Na komputerze kwantowym obliczenie każdego elementu macierzowego można wykonać za pomocą dowolnego algorytmu, który pozwala nam uzyskać nakrycie między stanami kwantowymi. W tej lekcji skupimy się na teście Hadamard. Zakładając, że ma wymiar , Hamiltonian rzutowany na tę podprzestrzeń będzie miał wymiary . Przy wystarczająco małym (ogólnie wystarcza do uzyskania zbieżności oszacowań wartości własnych) możemy łatwo zdiagonalizować rzutowany Hamiltonian klasycznie. Nie możemy jednak bezpośrednio zdiagonalizować z powodu nieortogonalności wektorów przestrzeni Krylova. Będziemy musieli zmierzyć ich nakrycia i skonstruować macierz
Pozwala to nam rozwiązać zagadnienie własne w przestrzeni nieortogonalnej (zwane również uogólnionym zagadnieniem własnym)
Oszacowania wartości własnych i stanów własnych można następnie uzyskać, analizując rozwiązania tego uogólnionego zagadnienia własnego. Na przykład oszacowanie energii stanu podstawowego uzyskuje się, biorąc najmniejszą wartość własną oraz stan podstawowy z odpowiadającego wektora własnego . Współczynniki w określają wkład różnych wektorów, które rozpinają .
Uogólnione zagadnienie własne
Dlaczego nie możemy po prostu zdiagonalizować ? Ponieważ zawiera informacje o geometrii bazy Krylova (która jest nieortogonalna poza bardzo szczególnymi przypadkami), samo nie opisuje rzutu pełnego Hamiltonianu, więc jego wartości własne nie mają szczególnego związku z wartościami własnymi pełnego Hamiltonianu — mogłyby to być dowolne losowe wartości. Rozwiązanie uogólnionego zagadnienia własnego jest wymagane, aby uzyskać przybliżone wartości własne i wektory własne odpowiadające rzutowi pełnego Hamiltonianu na przestrzeń Krylova.
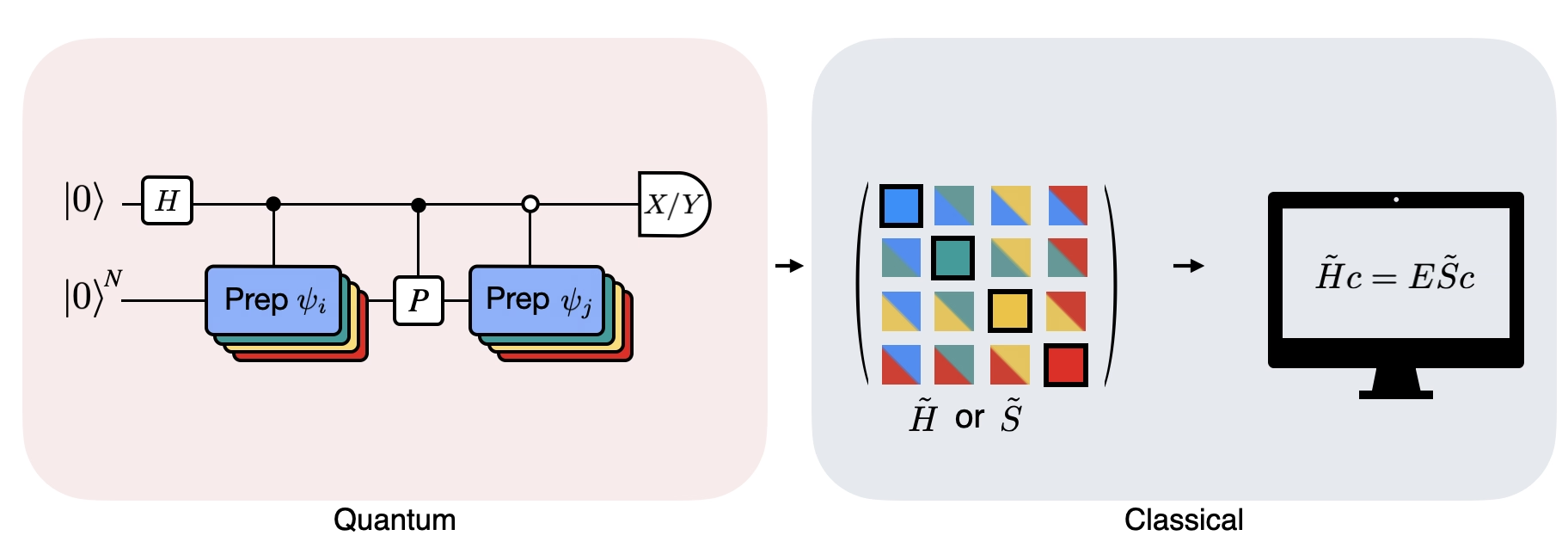
Rysunek pokazuje schematyczną reprezentację obwodu zmodyfikowanego testu Hadamard, metody stosowanej do obliczania nakrycia między różnymi stanami kwantowymi. Dla każdego elementu macierzowego wykonywany jest test Hadamard między stanami , . Zostało to wyróżnione na rysunku za pomocą schematu kolorów dla elementów macierzowych oraz odpowiadających im operacji , . W związku z tym, aby obliczyć wszystkie elementy macierzowe rzutowanego Hamiltonianu , wymagany jest zestaw testów Hadamard dla wszystkich możliwych kombinacji wektorów przestrzeni Krylova. Górna linia w obwodzie testu Hadamard jest kubitem pomocniczym (ancilla), który jest mierzony w bazie X lub Y, a jego wartość oczekiwana określa wartość nakrycia między stanami. Dolna linia reprezentuje wszystkie kubity Hamiltonianu systemu. Operacja przygotowuje kubit systemowy w stanie sterowany stanem kubitu ancilla (analogicznie dla ), a operacja reprezentuje dekompozycję Pauli Hamiltonianu systemu . Implementacja tego na komputerze kwantowym jest pokazana bardziej szczegółowo poniżej.
4. Kwantowa diagonalizacja Krylova na komputerze kwantowym
Zaimplementujemy teraz kwantową diagonalizację Krylova na prawdziwym komputerze kwantowym. Zacznijmy od zaimportowania kilku przydatnych pakietów.
import numpy as np
import scipy as sp
import matplotlib.pylab as plt
from typing import Union, List
import warnings
from qiskit.quantum_info import SparsePauliOp, Pauli
from qiskit.circuit import Parameter
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit.library import PauliEvolutionGate
from qiskit.synthesis import LieTrotter
# from qiskit.providers.fake_provider import Fake20QV1
from qiskit_ibm_runtime import QiskitRuntimeService, EstimatorV2 as Estimator, Batch
import itertools as it
warnings.filterwarnings("ignore")
Definiujemy poniższą funkcję, aby rozwiązać uogólnione zagadnienie własne, które właśnie wyjaśniliśmy powyżej.
def solve_regularized_gen_eig(
h: np.ndarray,
s: np.ndarray,
threshold: float,
k: int = 1,
return_dimn: bool = False,
) -> Union[float, List[float]]:
"""
Method for solving the generalized eigenvalue problem with regularization
Args:
h (numpy.ndarray):
The effective representation of the matrix in our Krylov subspace
s (numpy.ndarray):
The matrix of overlaps between vectors of our Krylov subspace
threshold (float):
Cut-off value for the eigenvalue of s
k (int):
Number of eigenvalues to return
return_dimn (bool):
Whether to return the size of the regularized subspace
Returns:
lowest k-eigenvalue(s) that are the solution of the regularized generalized eigenvalue problem
"""
s_vals, s_vecs = sp.linalg.eigh(s)
s_vecs = s_vecs.T
good_vecs = np.array([vec for val, vec in zip(s_vals, s_vecs) if val > threshold])
h_reg = good_vecs.conj() @ h @ good_vecs.T
s_reg = good_vecs.conj() @ s @ good_vecs.T
if k == 1:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][0], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][0]
else:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][:k], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][:k]
Przynajmniej we wstępnym benchmarkingu przydatne jest poznanie dokładnego rozwiązania klasycznego, aby sprawdzić zachowanie zbieżności. Poniższa funkcja oblicza energię stanu podstawowego Hamiltonianu, przyjmując jako argumenty Hamiltonian oraz liczbę kubitów.
def single_particle_gs(H_op, n_qubits):
"""
Find the ground state of the single particle(excitation) sector
"""
H_x = []
for p, coeff in H_op.to_list():
H_x.append(set([i for i, v in enumerate(Pauli(p).x) if v]))
H_z = []
for p, coeff in H_op.to_list():
H_z.append(set([i for i, v in enumerate(Pauli(p).z) if v]))
H_c = H_op.coeffs
print("n_sys_qubits", n_qubits)
n_exc = 1
sub_dimn = int(sp.special.comb(n_qubits + 1, n_exc))
print("n_exc", n_exc, ", subspace dimension", sub_dimn)
few_particle_H = np.zeros((sub_dimn, sub_dimn), dtype=complex)
sparse_vecs = [
set(vec) for vec in it.combinations(range(n_qubits + 1), r=n_exc)
] # list all of the possible sets of n_exc indices of 1s in n_exc-particle states
m = 0
for i, i_set in enumerate(sparse_vecs):
for j, j_set in enumerate(sparse_vecs):
m += 1
if len(i_set.symmetric_difference(j_set)) <= 2:
for p_x, p_z, coeff in zip(H_x, H_z, H_c):
if i_set.symmetric_difference(j_set) == p_x:
sgn = ((-1j) ** len(p_x.intersection(p_z))) * (
(-1) ** len(i_set.intersection(p_z))
)
else:
sgn = 0
few_particle_H[i, j] += sgn * coeff
gs_en = min(np.linalg.eigvalsh(few_particle_H))
print("single particle ground state energy: ", gs_en)
return gs_en
4.1 Krok 1: Mapowanie problemu na obwody i operatory kwantowe
Teraz zdefiniujemy Hamiltonian. Różni się on od powyższej funkcji tym, że powyższa funkcja przyjmuje Hamiltonian jako argument i zwraca jedynie stan podstawowy, i robi to klasycznie. Ten Hamiltonian, który definiujemy tutaj, określa poziomy energetyczne wszystkich stanów własnych energii, a ten Hamiltonian może być skonstruowany przy użyciu operatorów Pauli i zaimplementowany na komputerze kwantowym.
Wybieramy Hamiltonian odpowiadający łańcuchowi spinów, które mogą mieć dowolną orientację w przestrzeni, zwanym „łańcuchem Heisenberga”. Zakładamy, że -ty spin może być pod wpływem swoich najbliższych sąsiadów (spinów -tego i -tego), ale nie dalszych. Dopuszczamy również możliwość, że oddziaływanie między spinami jest różne, gdy spiny są skierowane wzdłuż różnych osi. Ta asymetria powstaje czasami, na przykład, z powodu struktury sieci krystalicznej, w której spiny są osadzone.
# Define problem Hamiltonian.
n_qubits = 10
# coupling strength for XX, YY, and ZZ interactions
JX = 1
JY = 3
JZ = 2
# Define the Hamiltonian:
H_int = [["I"] * n_qubits for _ in range(3 * (n_qubits - 1))]
for i in range(n_qubits - 1):
H_int[i][i] = "Z"
H_int[i][i + 1] = "Z"
for i in range(n_qubits - 1):
H_int[n_qubits - 1 + i][i] = "X"
H_int[n_qubits - 1 + i][i + 1] = "X"
for i in range(n_qubits - 1):
H_int[2 * (n_qubits - 1) + i][i] = "Y"
H_int[2 * (n_qubits - 1) + i][i + 1] = "Y"
H_int = ["".join(term) for term in H_int]
H_tot = [
(term, JZ)
if term.count("Z") == 2
else (term, JY)
if term.count("Y") == 2
else (term, JX)
for term in H_int
]
# Get operator
H_op = SparsePauliOp.from_list(H_tot)
print(H_tot)
[('ZZIIIIIIII', 2), ('IZZIIIIIII', 2), ('IIZZIIIIII', 2), ('IIIZZIIIII', 2), ('IIIIZZIIII', 2), ('IIIIIZZIII', 2), ('IIIIIIZZII', 2), ('IIIIIIIZZI', 2), ('IIIIIIIIZZ', 2), ('XXIIIIIIII', 1), ('IXXIIIIIII', 1), ('IIXXIIIIII', 1), ('IIIXXIIIII', 1), ('IIIIXXIIII', 1), ('IIIIIXXIII', 1), ('IIIIIIXXII', 1), ('IIIIIIIXXI', 1), ('IIIIIIIIXX', 1), ('YYIIIIIIII', 3), ('IYYIIIIIII', 3), ('IIYYIIIIII', 3), ('IIIYYIIIII', 3), ('IIIIYYIIII', 3), ('IIIIIYYIII', 3), ('IIIIIIYYII', 3), ('IIIIIIIYYI', 3), ('IIIIIIIIYY', 3)]
Kod poniżej ogranicza Hamiltonian do stanów jednocząstkowych oraz używa normy spektralnej, aby ustalić dobry rozmiar naszego kroku czasowego . Heurystycznie dobieramy wartość kroku czasowego dt (na podstawie górnych ograniczeń normy Hamiltonianu). Odniesienie [9] pokazało, że wystarczająco mały krok czasowy to i że do pewnego stopnia lepiej jest niedoszacować tę wartość niż ją przeszacować, ponieważ przeszacowanie może pozwolić wkładom ze stanów wysokoenergetycznych zepsuć nawet optymalny stan w przestrzeni Krylov. Z drugiej strony, zbyt mały wybór prowadzi do gorszego uwarunkowania podprzestrzeni Krylov, ponieważ wektory bazowe Krylov mniej różnią się między kolejnymi krokami czasowymi.
# Get Hamiltonian restricted to single-particle states
single_particle_H = np.zeros((n_qubits, n_qubits))
for i in range(n_qubits):
for j in range(i + 1):
for p, coeff in H_op.to_list():
p_x = Pauli(p).x
p_z = Pauli(p).z
if all(p_x[k] == ((i == k) + (j == k)) % 2 for k in range(n_qubits)):
sgn = ((-1j) ** sum(p_z[k] and p_x[k] for k in range(n_qubits))) * (
(-1) ** p_z[i]
)
else:
sgn = 0
single_particle_H[i, j] += sgn * coeff
for i in range(n_qubits):
for j in range(i + 1, n_qubits):
single_particle_H[i, j] = np.conj(single_particle_H[j, i])
# Set dt according to spectral norm
dt = np.pi / np.linalg.norm(single_particle_H, ord=2)
dt
np.float64(0.17453292519943295)
Określamy liczbę kroków Trottera do wykorzystania w ewolucji czasowej. Określamy również maksymalny wymiar Krylov wynoszący 4. Ten wymiar Krylov nie jest wystarczający dla realistycznych zastosowań, ale jest wystarczający dla tego przykładu. Ponadto zbadamy zbieżność przy jeszcze mniejszych wymiarach. W późniejszych lekcjach zbadamy metody, które pozwalają nam skalować i rzutować nasze Hamiltoniany na większe podprzestrzenie.
# Set parameters for quantum Krylov algorithm
krylov_dim = 4 # size of krylov subspace
num_trotter_steps = 4
dt_circ = dt / num_trotter_steps
Przygotowanie stanu
Wybierz stan referencyjny , który ma pewne pokrycie ze stanem podstawowym. Dla tego Hamiltonianu jako stan referencyjny używamy stanu ze wzbudzeniem na środkowym kubicie .
qc_state_prep = QuantumCircuit(n_qubits)
qc_state_prep.x(int(n_qubits / 2) + 1)
qc_state_prep.draw("mpl", scale=0.5)

Ewolucja czasowa
Możemy zrealizować operator ewolucji czasowej generowany przez dany Hamiltonian: za pomocą przybliżenia Liego-Trottera. Dla uproszczenia używamy wbudowanej PauliEvolutionGate w obwodzie ewolucji czasowej. Ogólna składnia wygląda tak.
t = Parameter("t")
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=t, synthesis=LieTrotter(reps=num_trotter_steps)
)
qr = QuantumRegister(n_qubits)
qc_evol = QuantumCircuit(qr)
qc_evol.append(evol_gate, qargs=qr)
<qiskit.circuit.instructionset.InstructionSet at 0x7ccaa4664250>
Wersji tego użyjemy poniżej w teście Hadamarda, ale wykonując kroki o czasie .
Test Hadamarda
Przypomnijmy, że chcemy obliczyć elementy macierzowe zarówno , jak i macierzy Grama przy użyciu testu Hadamarda. Przyjrzyjmy się, jak to działa w tym kontekście, skupiając się najpierw na konstrukcji Cały proces jest przedstawiony graficznie poniżej. Warstwy kolorowych bloków przygotowania stanu służą jako przypomnienie, że proces ten jest wykonywany dla wszystkich kombinacji i w naszej podprzestrzeni.
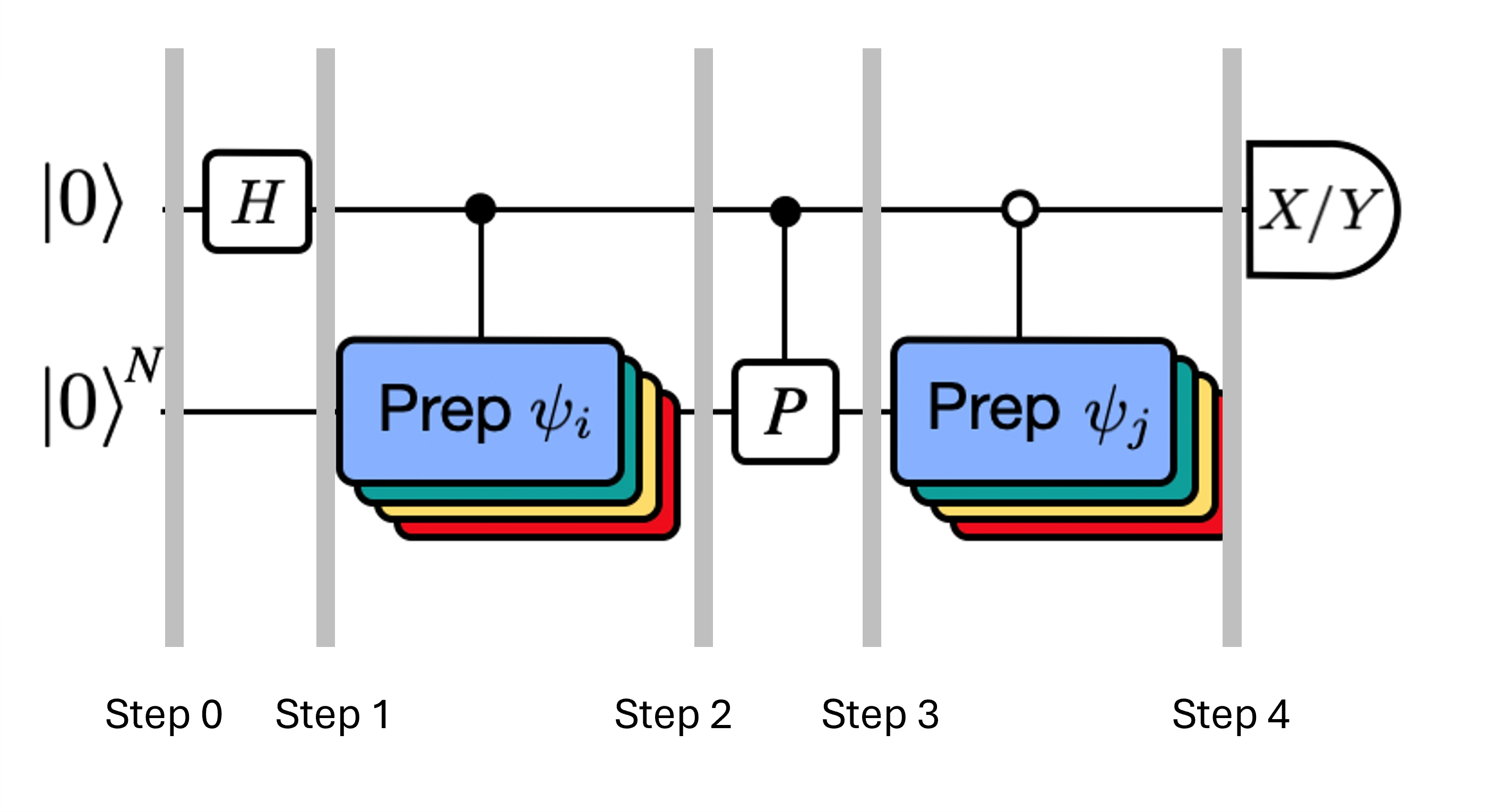
Stany układu w zaznaczonych krokach to:
Tutaj jest składnikiem Pauliego w rozkładzie Hamiltonianu (zauważ, że nie może to być kombinacja liniowa wielu przemiennych składników Pauliego, ponieważ nie byłoby to unitarne — grupowanie jest możliwe przy użyciu innej konstrukcji, którą pokażemy później). , to operacje kontrolowane, które przygotowują wektory , unitarnej przestrzeni Krylov, gdzie . Zastosowanie pomiarów i do tego obwodu pozwala obliczyć odpowiednio części rzeczywistą i urojoną elementów macierzowych, których potrzebujemy.
Zaczynając od Kroku 4 powyżej, zastosuj bramkę Hadamarda do zerowego kubitu.
Następnie zmierz lub .
Z tożsamości . Podobnie, pomiar daje
Dodając te kroki do ewolucji czasowej, którą wcześniej skonfigurowaliśmy, piszemy, co następuje.
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
## Create the time-evo op dagger circuit
evol_gate_d = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
evol_gate_d = evol_gate_d.inverse()
# Put pieces together
qc_reg = QuantumRegister(n_qubits)
qc_temp = QuantumCircuit(qc_reg)
qc_temp.compose(qc_state_prep, inplace=True)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate, qargs=qc_reg)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate_d, qargs=qc_reg)
qc_temp.compose(qc_state_prep.inverse(), inplace=True)
# Create controlled version of the circuit
controlled_U = qc_temp.to_gate().control(1)
# Create hadamard test circuit for real part
qr = QuantumRegister(n_qubits + 1)
qc_real = QuantumCircuit(qr)
qc_real.h(0)
qc_real.append(controlled_U, list(range(n_qubits + 1)))
qc_real.h(0)
print("Circuit for calculating the real part of the overlap in S via Hadamard test")
qc_real.draw("mpl", fold=-1, scale=0.5)
Circuit for calculating the real part of the overlap in S via Hadamard test

Ostrzegaliśmy już o głębokości obwodów Trottera. Wykonanie testu Hadamarda w tych warunkach może skutkować jeszcze głębszym obwodem, zwłaszcza po dekompozycji na bramki natywne. Głębokość ta wzrośnie jeszcze bardziej, jeśli uwzględnimy topologię urządzenia. Dlatego przed wykorzystaniem czasu na komputerze kwantowym dobrze jest sprawdzić głębokość 2-kubitową naszego obwodu.
print(
"Number of layers of 2Q operations",
qc_real.decompose(reps=2).depth(lambda x: x[0].num_qubits == 2),
)
Number of layers of 2Q operations 14401
Obwód o takiej głębokości nie może zwrócić użytecznych wyników na współczesnych komputerach kwantowych. Jeśli mamy skonstruować i potrzebujemy lepszego sposobu. To jest powód wprowadzenia poniżej efektywnego testu Hadamarda.
4. 2 Krok 2. Optymalizacja obwodów i operatorów pod docelowy sprzęt
Efektywny test Hadamarda
Możemy zoptymalizować głębokie obwody testu Hadamarda, które uzyskaliśmy, wprowadzając pewne przybliżenia i opierając się na pewnych założeniach dotyczących modelowego Hamiltonianu. Rozważmy na przykład poniższy obwód dla testu Hadamarda:
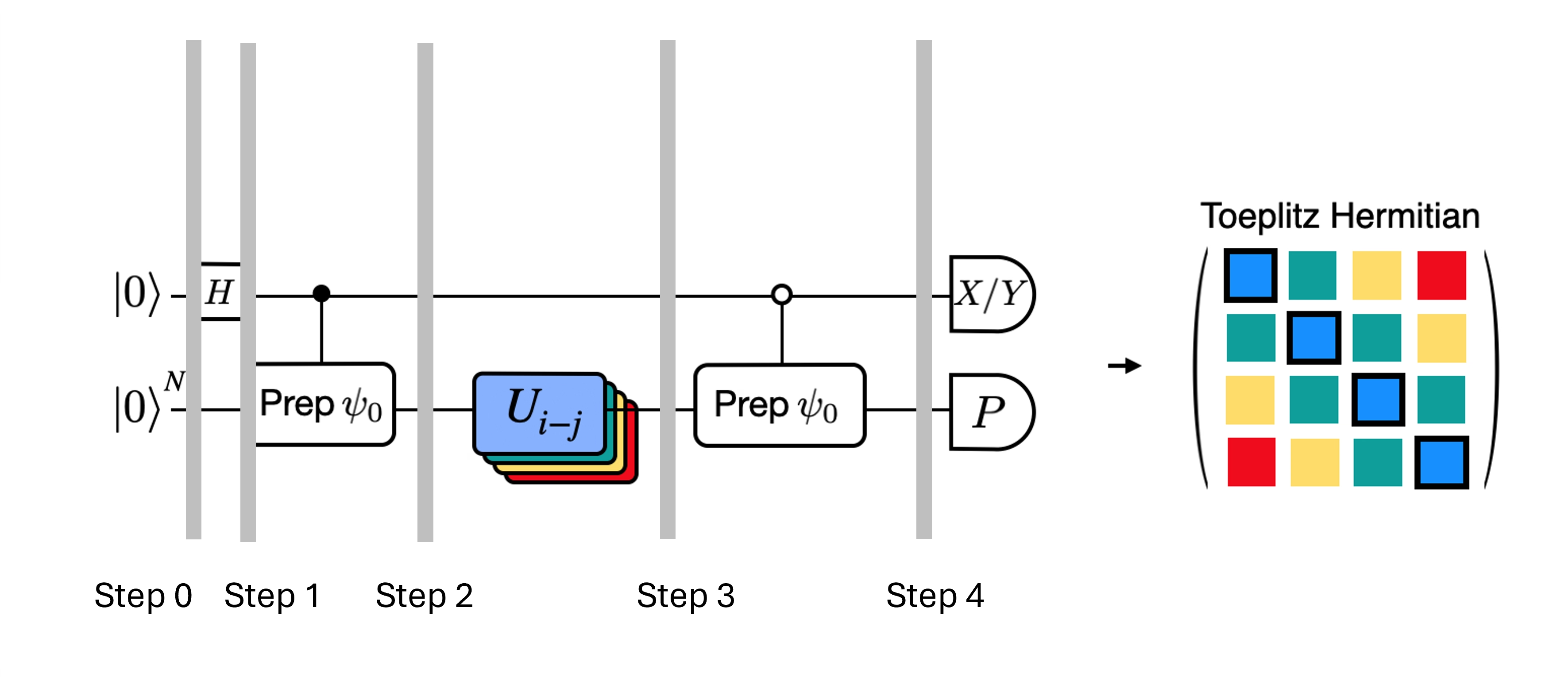
Załóżmy, że potrafimy klasycznie obliczyć , wartość własną pod Hamiltonianem . Jest to spełnione, gdy Hamiltonian zachowuje symetrię U(1). Choć może się to wydawać silnym założeniem, istnieje wiele przypadków, w których można bezpiecznie założyć, że istnieje stan próżni (w tym przypadku odwzorowywany na stan ), który nie jest zmieniany przez działanie Hamiltonianu. Jest to prawdziwe na przykład dla Hamiltonianów chemicznych opisujących stabilną cząsteczkę (gdzie liczba elektronów jest zachowana). Mając daną bramkę , która przygotowuje pożądany stan referencyjny , na przykład, aby przygotować stan HF dla chemii byłoby iloczynem jednokubitowych bramek NOT, więc kontrolowane jest po prostu iloczynem bramek CNOT. Wtedy powyższy obwód realizuje następujący stan przed pomiarem:
gdzie skorzystaliśmy z klasycznie symulowanego przesunięcia fazowego przechodząc od kroku 2 do 3. Zatem wartości oczekiwane wynoszą
Korzystając z tych założeń, udało nam się zapisać wartości oczekiwane interesujących nas operatorów przy mniejszej liczbie operacji kontrolowanych. W rzeczywistości musimy zaimplementować jedynie kontrolowane przygotowanie stanu , a nie kontrolowane ewolucje czasowe. Przeformułowanie naszych obliczeń w powyższy sposób pozwoli nam znacznie zmniejszyć głębokość powstałych obwodów.
Zauważ, że jako bonus, ponieważ operator Pauli pojawia się teraz jako pomiar na końcu obwodu, a nie jako bramka kontrolowana w środku, może być mierzony razem z innymi przemiennymi operatorami Pauli, jak w dekompozycji podanej powyżej.
Dekompozycja operatora ewolucji czasowej za pomocą dekompozycji Trottera
Zamiast implementować operator ewolucji czasowej dokładnie, możemy użyć dekompozycji Trottera, aby zaimplementować jego przybliżenie. Kilkukrotne powtórzenie dekompozycji Trottera pewnego rzędu daje nam dalsze zmniejszenie błędu wprowadzonego przez to przybliżenie. W dalszej części bezpośrednio budujemy implementację Trottera w najbardziej efektywny sposób dla grafu oddziaływań rozważanego przez nas Hamiltonianu (oddziaływania jedynie najbliższych sąsiadów). W praktyce wstawiamy obroty Pauli , , z siłami sprzężenia , , oraz parametryzowanym kątem , które odpowiadają przybliżonej implementacji . Biorąc pod uwagę różnicę w definicji obrotów Pauli i ewolucji czasowej, którą próbujemy zaimplementować, będziemy musieli użyć parametru , aby osiągnąć ewolucję czasową o . Ponadto odwracamy kolejność operacji dla nieparzystej liczby powtórzeń kroków Trottera, co jest funkcjonalnie równoważne, ale pozwala na syntezę sąsiednich operacji w pojedynczym unitarnym . Daje to znacznie płytszy obwód niż ten uzyskany za pomocą ogólnej funkcjonalności PauliEvolutionGate().
t = Parameter("t")
# Create instruction for rotation about XX+YY-ZZ:
Rxyz_circ = QuantumCircuit(2)
Rxyz_circ.rxx(2 * JX * t, 0, 1)
Rxyz_circ.ryy(2 * JY * t, 0, 1)
Rxyz_circ.rzz(2 * JZ * t, 0, 1)
Rxyz_instr = Rxyz_circ.to_instruction(label="R J_x XX + J_y YY + J_z ZZ")
interaction_list = [
[[i, i + 1] for i in range(0, n_qubits - 1, 2)],
[[i, i + 1] for i in range(1, n_qubits - 1, 2)],
] # linear chain
qr = QuantumRegister(n_qubits)
trotter_step_circ = QuantumCircuit(qr)
for i, color in enumerate(interaction_list):
for interaction in color:
trotter_step_circ.append(Rxyz_instr, interaction)
if i < len(interaction_list) - 1:
trotter_step_circ.barrier()
reverse_trotter_step_circ = trotter_step_circ.reverse_ops()
qc_evol = QuantumCircuit(qr)
for step in range(num_trotter_steps):
if step % 2 == 0:
qc_evol = qc_evol.compose(trotter_step_circ)
else:
qc_evol = qc_evol.compose(reverse_trotter_step_circ)
qc_evol.decompose().draw("mpl", fold=-1, scale=0.5)
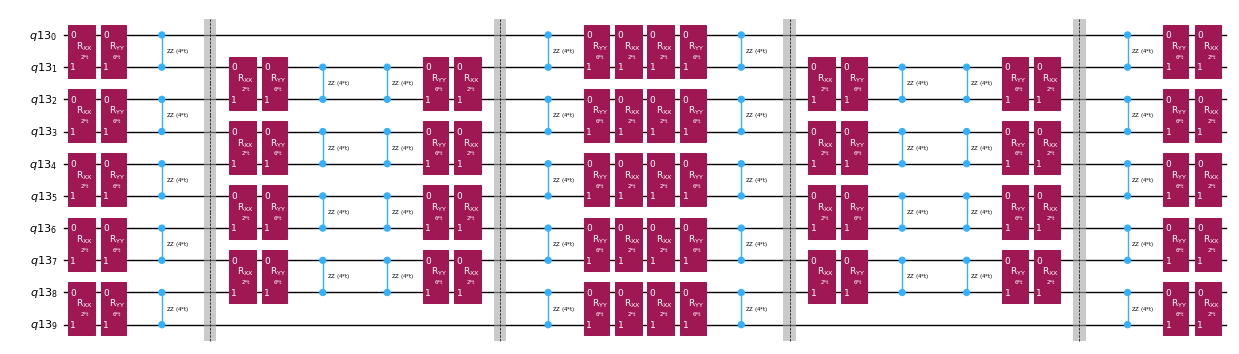
Ponownie przygotowujemy stan początkowy dla tego efektywnego testu Hadamarda.
control = 0
excitation = int(n_qubits / 2) + 1
controlled_state_prep = QuantumCircuit(n_qubits + 1)
controlled_state_prep.cx(control, excitation)
controlled_state_prep.draw("mpl", fold=-1, scale=0.5)

Szablonowe obwody do obliczania elementów macierzowych i za pomocą testu Hadamarda
Jedyną różnicą pomiędzy obwodami używanymi w teście Hadamarda będzie faza w operatorze ewolucji czasowej oraz mierzone obserwable. Dlatego możemy przygotować obwód szablonowy, który reprezentuje ogólny obwód dla testu Hadamarda, z miejscami zastępczymi na bramki zależące od operatora ewolucji czasowej.
# Parameters for the template circuits
parameters = []
for idx in range(1, krylov_dim):
parameters.append(dt_circ * (idx))
# Create modified hadamard test circuit
qr = QuantumRegister(n_qubits + 1)
qc = QuantumCircuit(qr)
qc.h(0)
qc.compose(controlled_state_prep, list(range(n_qubits + 1)), inplace=True)
qc.barrier()
qc.compose(qc_evol, list(range(1, n_qubits + 1)), inplace=True)
qc.barrier()
qc.x(0)
qc.compose(controlled_state_prep.inverse(), list(range(n_qubits + 1)), inplace=True)
qc.x(0)
qc.decompose().draw("mpl", fold=-1)
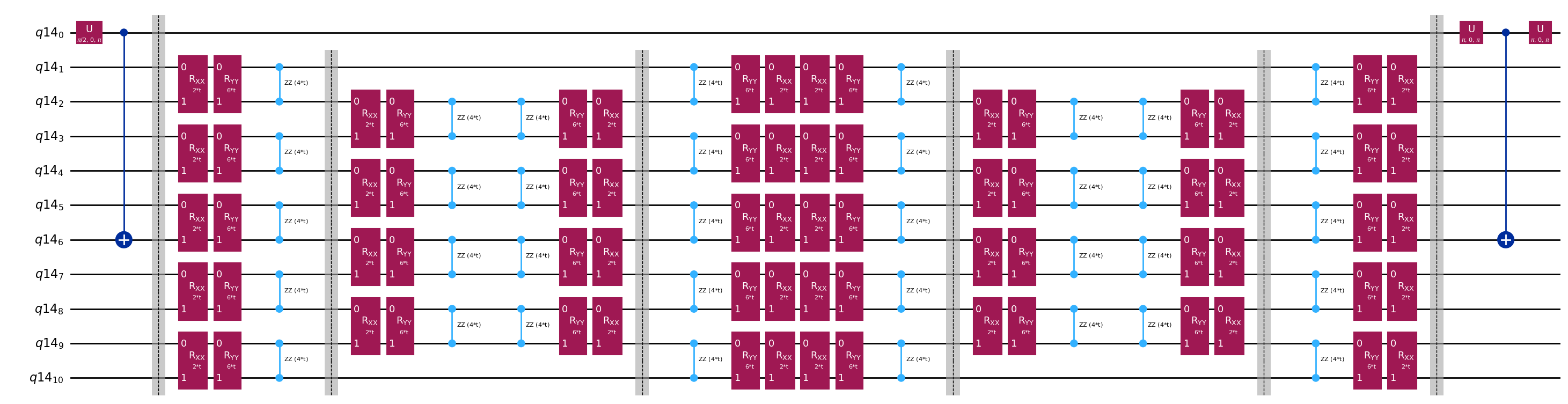
print(
"The optimized circuit has 2Q gates depth: ",
qc.decompose().decompose().depth(lambda x: x[0].num_qubits == 2),
)
The optimized circuit has 2Q gates depth: 50
Głębokość ta jest znacznie zmniejszona w porównaniu z oryginalnym testem Hadamarda. Głębokość ta jest wykonalna na współczesnych komputerach kwantowych, choć wciąż jest dość duża. Aby uzyskać użyteczne wyniki, będziemy musieli zastosować najnowocześniejsze techniki mitygacji błędów.
Wybierz backend, na którym uruchomisz nasze obliczenia kwantowe metodą Krylov, abyśmy mogli transpilować nasz obwód do uruchomienia na tym komputerze kwantowym.
# Use the least-busy backend or specify a quantum computer using the syntax commented out below.
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
# Or you may choose a specify backend and channel if necessary for your workflow.
# service = QiskitRuntimeService(channel="ibm_quantum_platform")
# backend = service.backend("ibm_fez")
Teraz transpilujemy nasze obwody i operatory.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
basis_gates = list(target.operation_names)
pm = generate_preset_pass_manager(
optimization_level=3, backend=backend, basis_gates=basis_gates
)
qc_trans = pm.run(qc)
print(qc_trans.depth(lambda x: x[0].num_qubits == 2))
print(qc_trans.count_ops())
qc_trans.draw("mpl", fold=-1, idle_wires=False, scale=0.5)
36
OrderedDict([('rz', 410), ('sx', 361), ('cz', 156), ('x', 18), ('barrier', 6)])

Po optymalizacji nasza stranspilowana głębokość dwukubitowa jest dodatkowo zredukowana.
4.3 Krok 3. Wykonanie przy użyciu prymitywu Qiskit Runtime
Tworzymy teraz PUB-y do wykonania z użyciem Estimatora.
# Define observables to measure for S
observable_S_real = "I" * (n_qubits) + "X"
observable_S_imag = "I" * (n_qubits) + "Y"
observable_op_real = SparsePauliOp(
observable_S_real
) # define a sparse pauli operator for the observable
observable_op_imag = SparsePauliOp(observable_S_imag)
layout = qc_trans.layout # get layout of transpiled circuit
observable_op_real = observable_op_real.apply_layout(
layout
) # apply physical layout to the observable
observable_op_imag = observable_op_imag.apply_layout(layout)
observable_S_real = (
observable_op_real.paulis.to_labels()
) # get the label of the physical observable
observable_S_imag = observable_op_imag.paulis.to_labels()
observables_S = [[observable_S_real], [observable_S_imag]]
# Define observables to measure for H
# Hamiltonian terms to measure
observable_list = []
for pauli, coeff in zip(H_op.paulis, H_op.coeffs):
# print(pauli)
observable_H_real = pauli[::-1].to_label() + "X"
observable_H_imag = pauli[::-1].to_label() + "Y"
observable_list.append([observable_H_real])
observable_list.append([observable_H_imag])
layout = qc_trans.layout
observable_trans_list = []
for observable in observable_list:
observable_op = SparsePauliOp(observable)
observable_op = observable_op.apply_layout(layout)
observable_trans_list.append([observable_op.paulis.to_labels()])
observables_H = observable_trans_list
# Define a sweep over parameter values
params = np.vstack(parameters).T
# Estimate the expectation value for all combinations of
# observables and parameter values, where the pub result will have
# shape (# observables, # parameter values).
pub = (qc_trans, observables_S + observables_H, params)
Obwody dla są obliczalne klasycznie. Wykonujemy to, zanim przejdziemy do przypadku z wykorzystaniem komputera kwantowego.
from qiskit.quantum_info import StabilizerState, Pauli
qc_cliff = qc.assign_parameters({t: 0})
# Get expectation values from experiment
S_expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "X")
)
S_expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "Y")
)
# Get expectation values
S_expval = S_expval_real + 1j * S_expval_imag
H_expval = 0
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Get expectation values from experiment
expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "X")
)
expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "Y")
)
expval = expval_real + 1j * expval_imag
# Fill-in matrix elements
H_expval += coeff * expval
print(H_expval)
(10+0j)
Chociaż udało nam się zredukować głębokość bramek o rzędy wielkości przy użyciu wydajnego testu Hadamarda, głębokość jest nadal wystarczająca, aby wymagać najnowocześniejszej mitygacji błędów. Poniżej określamy atrybuty stosowanej mitygacji. Wszystkie używane metody są ważne, ale warto szczególnie wyróżnić probabilistyczne wzmacnianie błędów (PEA). Ta potężna technika wiąże się ze znacznym narzutem kwantowym. Wykonywane tu obliczenia mogą zająć 20 minut lub więcej na rzeczywistym komputerze kwantowym. Możesz poeksperymentować z parametrami poniżej, aby zwiększyć lub zmniejszyć dokładność oraz wynikający z niej narzut. Domyślne ustawienia poniżej zapewniają wyniki o wysokiej wierności.
# Experiment options
num_randomizations = 300
num_randomizations_learning = 20
max_batch_circuits = 20
shots_per_randomization = 100
learning_pair_depths = [0, 4, 24]
noise_factors = [1, 1.3, 1.6]
# Base option formatting
options = {
# Builtin resilience settings for ZNE
"resilience": {
"measure_mitigation": True,
"zne_mitigation": True,
"zne": {"noise_factors": noise_factors},
# TREX noise learning configuration
"measure_noise_learning": {
"num_randomizations": num_randomizations_learning,
"shots_per_randomization": shots_per_randomization,
},
# PEA noise model configuration
"layer_noise_learning": {
"max_layers_to_learn": 10,
"layer_pair_depths": learning_pair_depths,
"shots_per_randomization": shots_per_randomization,
"num_randomizations": num_randomizations_learning,
},
},
# Randomization configuration
"twirling": {
"num_randomizations": num_randomizations,
"shots_per_randomization": shots_per_randomization,
"strategy": "all",
},
# Experimental settings for PEA method
"experimental": {
# # Just in case, disable any further qiskit transpilation not related to twirling / DD
# "skip_transpilation": True,
# Execution configuration
"execution": {
"max_pubs_per_batch_job": max_batch_circuits,
"fast_parametric_update": True,
},
# Error Mitigation configuration
"resilience": {
# ZNE Configuration
"zne": {
"amplifier": "pea",
"return_all_extrapolated": True,
"return_unextrapolated": True,
"extrapolated_noise_factors": [0] + noise_factors,
}
},
},
}
Na koniec uruchamiamy obwody dla i za pomocą Estimator.
# This job required 17 minutes of QPU time to run on a Heron r2 processor. This is only an estimate.
# Your execution time may vary.
with Batch(backend=backend) as batch:
# Estimator
estimator = Estimator(mode=batch, options=options)
job = estimator.run([pub], precision=1)
4.4 Krok 4. Przetwarzanie końcowe i analiza wyników
To, co otrzymaliśmy z komputera kwantowego, to poszczególne elementy macierzowe oraz komutujące grupy Paulich, które tworzą elementy macierzowe . Terminy te trzeba połączyć, aby odzyskać nasze macierze i móc rozwiązać uogólnione zagadnienie własne.
# Store the outputs as 'results'.
results = job.result()[0]
Oblicz efektywny Hamiltonian i macierze nakrywania
Najpierw oblicz fazę nagromadzoną przez stan podczas niesterowanej ewolucji czasowej
prefactors = [
np.exp(-1j * sum([c for p, c in H_op.to_list() if "Z" in p]) * i * dt)
for i in range(1, krylov_dim)
]
Gdy mamy już wyniki wykonań obwodów, możemy przeprowadzić przetwarzanie końcowe danych, aby obliczyć elementy macierzowe
# Assemble S, the overlap matrix of dimension D:
S_first_row = np.zeros(krylov_dim, dtype=complex)
S_first_row[0] = 1 + 0j
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[0][0][i] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[1][0][i] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
S_first_row[i + 1] += prefactors[i] * expval
S_first_row_list = S_first_row.tolist() # for saving purposes
S_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in it.product(range(krylov_dim), repeat=2):
if i >= j:
S_circ[j, i] = S_first_row[i - j]
else:
S_circ[j, i] = np.conj(S_first_row[j - i])
from sympy import Matrix
Matrix(S_circ)
Oraz elementy macierzowe
import itertools
# Assemble S, the overlap matrix of dimension D:
H_first_row = np.zeros(krylov_dim, dtype=complex)
H_first_row[0] = H_expval
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[2 + 2 * obs_idx][0][
i
] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[2 + 2 * obs_idx + 1][0][
i
] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
H_first_row[i + 1] += prefactors[i] * coeff * expval
H_first_row_list = H_first_row.tolist()
H_eff_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in itertools.product(range(krylov_dim), repeat=2):
if i >= j:
H_eff_circ[j, i] = H_first_row[i - j]
else:
H_eff_circ[j, i] = np.conj(H_first_row[j - i])
from sympy import Matrix
Matrix(H_eff_circ)
Ostatecznie możemy rozwiązać uogólnione zagadnienie własne dla :
i uzyskać oszacowanie energii stanu podstawowego
gnd_en_circ_est_list = []
for d in range(1, krylov_dim + 1):
# Solve generalized eigenvalue problem
gnd_en_circ_est = solve_regularized_gen_eig(
H_eff_circ[:d, :d], S_circ[:d, :d], threshold=1e-1
)
gnd_en_circ_est_list.append(gnd_en_circ_est)
print("The estimated ground state energy is: ", gnd_en_circ_est)
The estimated ground state energy is: 10.0
The estimated ground state energy is: 5.933953916292923
The estimated ground state energy is: 4.4101773995740645
The estimated ground state energy is: 3.921288588521255
Dla sektora jednocząstkowego możemy efektywnie obliczyć stan podstawowy tego sektora Hamiltonianu klasycznie
gs_en = single_particle_gs(H_op, n_qubits)
n_sys_qubits 10
n_exc 1 , subspace dimension 11
single particle ground state energy: 2.391547869638771
len(H_op)
27
plt.plot(
range(1, krylov_dim + 1),
gnd_en_circ_est_list,
color="blue",
linestyle="-.",
label="KQD estimate",
)
plt.plot(
range(1, krylov_dim + 1),
[gs_en] * krylov_dim,
color="red",
linestyle="-",
label="exact",
)
plt.xticks(range(1, krylov_dim + 1), range(1, krylov_dim + 1))
plt.legend()
plt.xlabel("Krylov space dimension")
plt.ylabel("Energy")
plt.title("Estimating Ground state energy with Krylov Quantum Diagonalization")
plt.show()
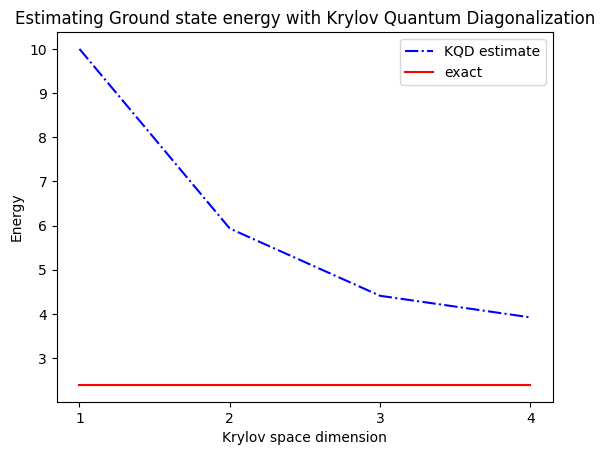
5. Dyskusja i rozszerzenie
Podsumowując, zaczynamy od stanu referencyjnego, a następnie ewoluujemy go przez różne okresy czasu, aby wygenerować unitarną podprzestrzeń Krylova. Rzutujemy nasz Hamiltonian na tę podprzestrzeń. Estymujemy również nakładania się wektorów podprzestrzeni. Na koniec klasycznie rozwiązujemy uogólniony problem własny o niższym wymiarze.
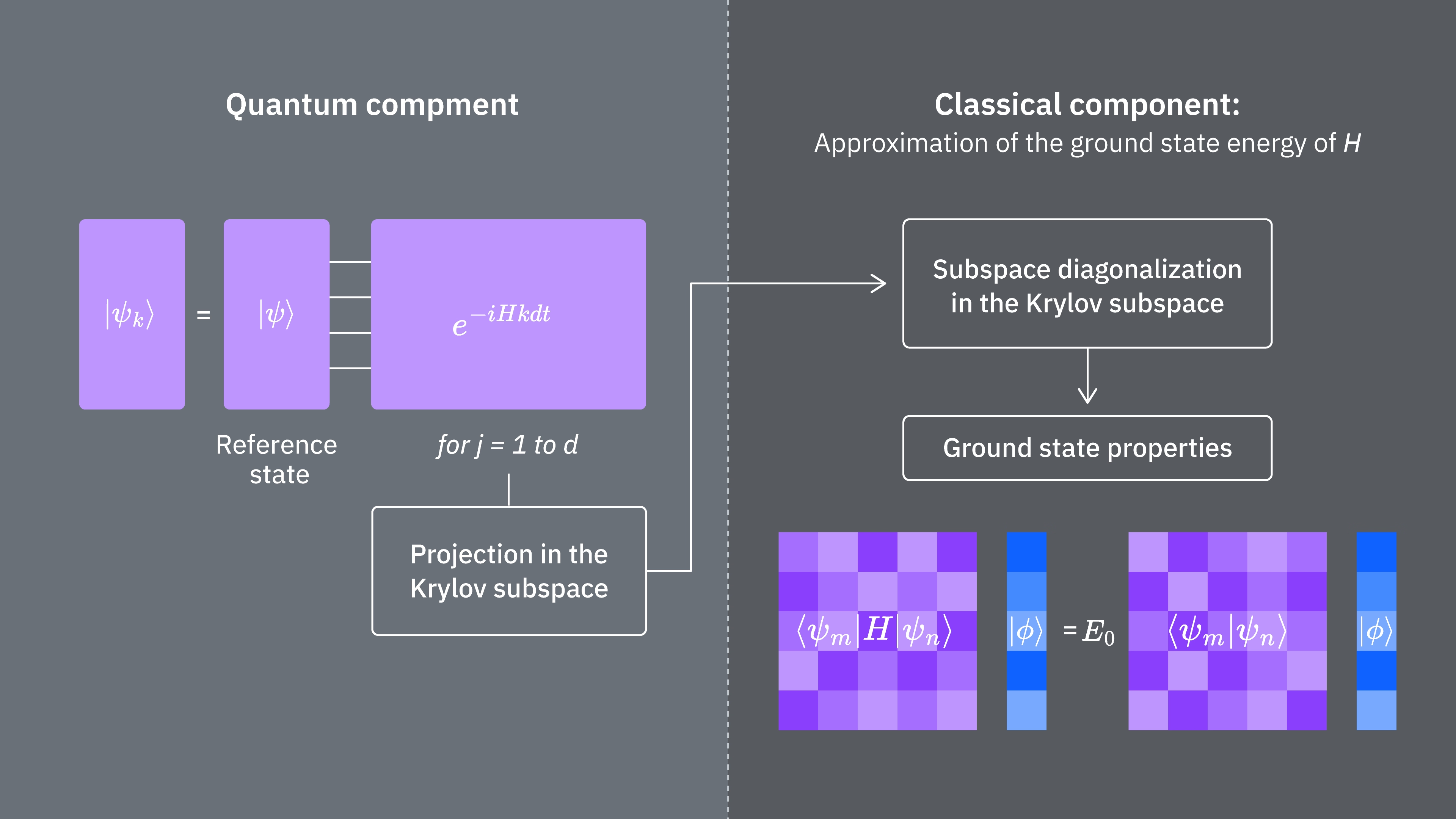
Porównajmy, co określa koszty obliczeniowe korzystania z techniki Krylova klasycznie i kwantowo-mechanicznie. Nie istnieją doskonałe analogie pomiędzy podejściem klasycznym a kwantowym dla wszystkich kroków. Poniższa tabela zbiera niektóre skalowania różnych kroków do rozważenia.
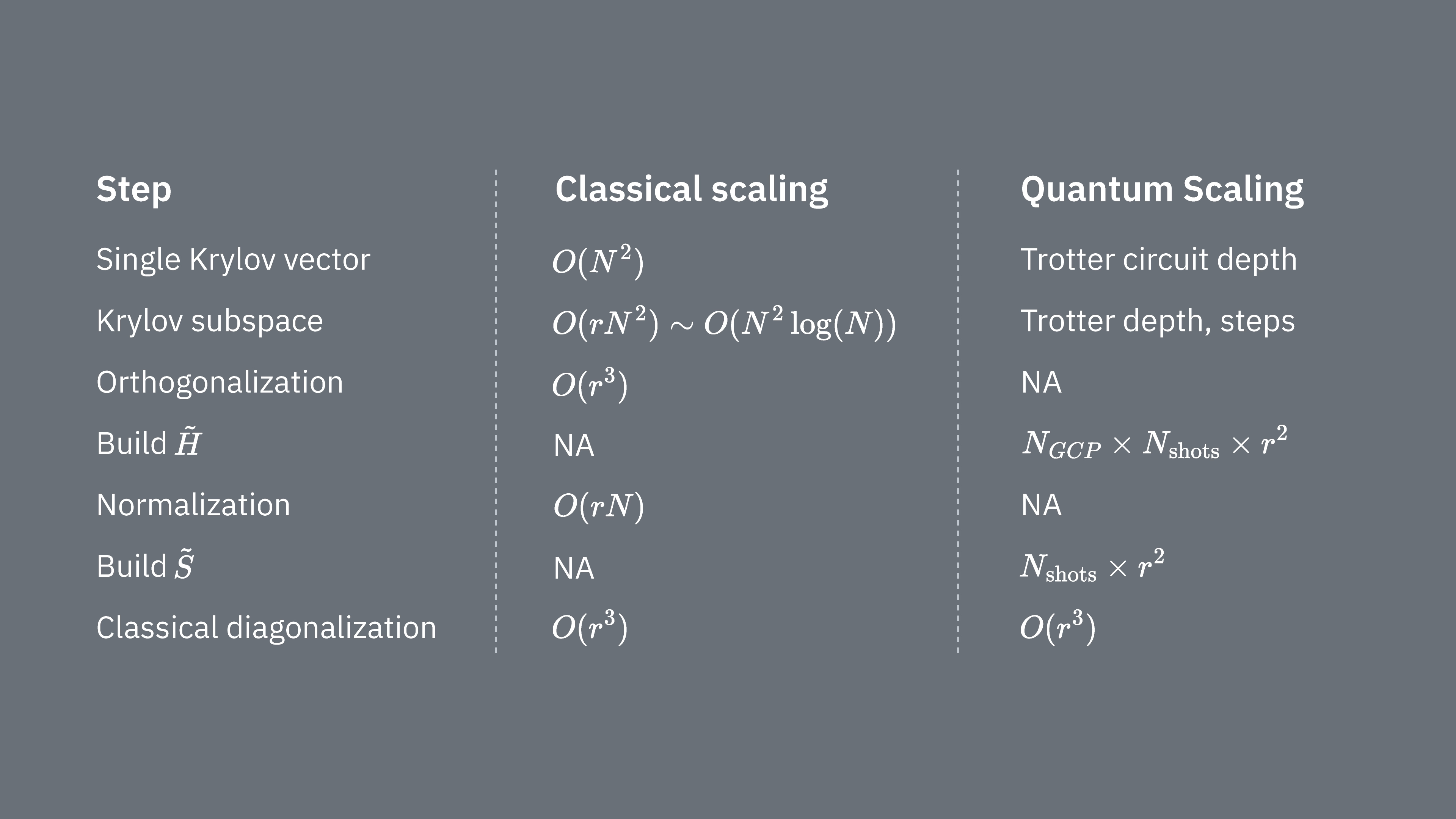
Przypomnijmy, że Hamiltoniany zazwyczaj mają człony, których nie można jednocześnie mierzyć (ponieważ nie komutują ze sobą). Sortujemy człony Hamiltonianu w grupy komutujących operatorów Pauli, które wszystkie mogą być mierzone jednocześnie, i możemy potrzebować wielu takich grup, aby uwzględnić wszystkie człony, które nie komutują ze sobą. Aby zbudować na komputerze kwantowym, wymagane są osobne pomiary dla każdej grupy komutujących ciągów Pauli w Hamiltonianie, a każdy z nich wymaga wielu strzałów. Musimy zrobić to dla różnych elementów macierzowych, odpowiadających kombinacjom różnych czynników ewolucji czasowej. Czasami istnieją sposoby, aby to zredukować, ale w tym zgrubnym ujęciu czas wymagany do tego skaluje się jak Elementy muszą być estymowane, co skaluje się jak . Wreszcie klasyczne rozwiązanie uogólnionego problemu własnego w rzutowanej przestrzeni zajmuje
Widzimy, że kwantowa diagonalizacja Krylova może być przydatna w przypadkach, gdy liczba komutujących grup Pauli w Hamiltonianie jest stosunkowo mała. Te zależności skalowania sugerują niektóre zastosowania, w których metoda Krylova może być użyteczna, i inne, w których prawdopodobnie nie będzie. Niektóre Hamiltoniany mają wysoką złożoność po odwzorowaniu na kubity, angażując wiele niekomutujących ciągów Pauli, które nie dają się łatwo podzielić na kilka grup komutujących. Często dotyczy to na przykład problemów chemii kwantowej. Ta złożoność stawia dwa główne wyzwania dla komputerów kwantowych bliskiej przyszłości:
- Estymacja każdego elementu staje się kosztowna obliczeniowo ze względu na dużą liczbę członów.
- Wymagane obwody Trottera stają się zbyt głębokie, aby były praktyczne.
Oba powyższe punkty będą mniej problematyczne, gdy komputery kwantowe osiągną odporność na błędy, ale muszą być brane pod uwagę w bliskiej perspektywie. Nawet systemy z „prostszymi” odwzorowaniami niż te w chemii kwantowej mogą doświadczać tych samych utrudnień, jeśli Hamiltoniany mają zbyt wiele niekomutujących członów. Metoda Krylova jest najbardziej użyteczna tam, gdzie Hamiltonian może być podzielony na stosunkowo niewiele komutujących grup Pauli i gdzie jest łatwe do implementacji w obwodach Trottera. Oba te warunki są spełnione, na przykład, dla wielu modeli sieciowych interesujących w fizyce. KQD jest szczególnie użyteczna, jeśli bardzo mało wiadomo o stanie podstawowym. Wynika to z jej wrodzonych gwarancji zbieżności i jej zastosowania w scenariuszach, w których alternatywne metody są nie do utrzymania z powodu niewystarczającej wiedzy o stanie podstawowym.
Chociaż KQD jest potężnym narzędziem, czasochłonne aspekty protokołu, w szczególności estymacja każdego elementu rzutowanego Hamiltonianu i nakładania się stanów Krylova, stanowią możliwości poprawy. Alternatywne podejście polega na wykorzystaniu metod Krylova w połączeniu z metodami opartymi na próbkowaniu, co jest tematem następnej lekcji.
6. Dodatki
Dodatek I: Podprzestrzeń Krylova z ewolucji w czasie rzeczywistym
Unitarna przestrzeń Krylova jest zdefiniowana jako
dla pewnego kroku czasowego , który określimy później. Tymczasowo załóżmy, że jest parzyste: wtedy definiujemy . Zauważmy, że gdy rzutujemy Hamiltonian na powyższą przestrzeń Krylova, jest on nierozróżnialny od przestrzeni Krylova
to znaczy gdzie wszystkie ewolucje w czasie są przesunięte wstecz o kroków czasowych. Powodem, dla którego jest nierozróżnialny, jest to, że elementy macierzowe
są niezmiennicze względem globalnych przesunięć czasu ewolucji, ponieważ ewolucje w czasie komutują z Hamiltonianem. Dla nieparzystego możemy użyć analizy dla .
Chcemy pokazać, że gdzieś w tej przestrzeni Krylova gwarantowane jest istnienie stanu niskoenergetycznego. Robimy to za pomocą następującego wyniku, wyprowadzonego z Twierdzenia 3.1 w [3]:
Twierdzenie 1: istnieje funkcja taka, że dla energii w zakresie spektralnym Hamiltonianu (to znaczy między energią stanu podstawowego a energią maksymalną)...
- dla wszystkich wartości , które leżą od , to znaczy jest wykładniczo tłumiona
- jest kombinacją liniową dla
Dowód podajemy poniżej, ale można go bezpiecznie pominąć, chyba że ktoś chce zrozumieć pełną, ścisłą argumentację. Na razie skupiamy się na implikacjach powyższego twierdzenia. Z własności 3 powyżej widzimy, że przesunięta przestrzeń Krylova powyżej zawiera stan . To jest nasz stan niskoenergetyczny. Aby zobaczyć dlaczego, zapiszmy w bazie własnej energii:
gdzie jest k-tym stanem własnym energii, a jest jego amplitudą w stanie początkowym . Wyrażone w ten sposób, jest dane przez
wykorzystując fakt, że możemy zastąpić przez , gdy działa na stan własny . Błąd energii tego stanu jest zatem równy
Aby przekształcić to w górne ograniczenie, które jest łatwiejsze do zrozumienia, najpierw rozdzielamy sumę w liczniku na człony z i człony z :
Możemy oszacować pierwszy człon od góry przez ,
gdzie pierwszy krok wynika stąd, że dla każdego w sumie, a drugi krok wynika stąd, że suma w liczniku jest podzbiorem sumy w mianowniku. Dla drugiego członu najpierw oszacowujemy mianownik od dołu przez , ponieważ : składając wszystko z powrotem, otrzymujemy
Aby uprościć to, co pozostało, zauważmy, że dla wszystkich tych , z definicji wiemy, że . Dodatkowo szacując od góry i szacując od góry , otrzymujemy
To zachodzi dla dowolnego , więc jeśli ustawimy równe naszemu docelowemu błędowi, to powyższe ograniczenie błędu zbiega do niego wykładniczo wraz z wymiarem Krylova . Zauważmy również, że jeśli , to człon faktycznie całkowicie znika w powyższym ograniczeniu.
Aby dopełnić argumentu, zauważamy najpierw, że powyższe jest jedynie błędem energii konkretnego stanu , a nie błędem energii stanu o najniższej energii w przestrzeni Krylova. Jednakże, zgodnie z zasadą wariacyjną (Rayleigha-Ritza), błąd energii stanu o najniższej energii w przestrzeni Krylova jest ograniczony od góry przez błąd energii dowolnego stanu w przestrzeni Krylova, więc powyższe jest również górnym ograniczeniem dla błędu energii stanu o najniższej energii, to znaczy wyniku algorytmu kwantowej diagonalizacji Krylova.
Podobną analizę jak powyższa można przeprowadzić, uwzględniając dodatkowo szum i procedurę progowania omówioną w notatniku. Zobacz [2] oraz [4] dla tej analizy.
Dodatek II: dowód Twierdzenia 1
Poniższe jest głównie wyprowadzone z [3], Twierdzenie 3.1: niech i niech będzie przestrzenią wielomianów resztowych (wielomianów, których wartość w 0 wynosi 1) stopnia co najwyżej . Rozwiązaniem
jest
a odpowiadająca minimalna wartość wynosi
Chcemy przekształcić to w funkcję, którą można naturalnie wyrazić za pomocą zespolonych funkcji wykładniczych, ponieważ są to rzeczywiste ewolucje w czasie, które generują kwantową przestrzeń Krylova. Aby to zrobić, wygodnie jest wprowadzić następujące przekształcenie energii z zakresu spektralnego Hamiltonianu na liczby z przedziału : definiujemy
gdzie jest takim krokiem czasowym, że . Zauważmy, że , a rośnie, gdy oddala się od .
Teraz, używając wielomianu z parametrami a, b, d ustawionymi na , oraz d = int(r/2), definiujemy funkcję:
gdzie jest energią stanu podstawowego. Możemy zobaczyć, wstawiając , że jest wielomianem trygonometrycznym stopnia , to znaczy kombinacją liniową dla . Ponadto z definicji powyżej mamy, że oraz dla dowolnego w zakresie spektralnym takiego, że mamy
Bibliografia:
[1] https://arxiv.org/abs/2407.14431
[2] https://arxiv.org/abs/1811.09025
[3] https://people.math.ethz.ch/~mhg/pub/biksm.pdf
[4] https://academic.oup.com/book/36426
[5] https://en.wikipedia.org/wiki/Krylov_subspace
[6] Krylov Subspace Methods: Principles and Analysis, Jörg Liesen, Zdenek Strakos https://academic.oup.com/book/36426
[7] Iterative Methods for Sparse Linear Systems" by Yousef Saad
[8] "MINRES-QLP: A Krylov Subspace Method for Indefinite or Singular Symmetric Systems" by Sou-Cheng Choi, Christopher Paige, and Michael Saunders (https://epubs.siam.org/doi/10.1137/100787921)
[9] Ethan N. Epperly, Lin Lin, and Yuji Nakatsukasa. "A theory of quantum subspace diagonalization". SIAM Journal on Matrix Analysis and Applications 43, 1263–1290 (2022).