Poprawa estymacji energii fermionowego modelu sieciowego przy użyciu SQD
W tym samouczku implementujemy wzorzec Qiskit pokazujący, jak przetwarzać po stronie klasycznej zaszumione próbki kwantowe w celu znalezienia przybliżenia stanu podstawowego fermionowego Hamiltonianu sieciowego, zwanego modelem Andersona z pojedynczą domieszką. Zastosujemy podejście oparte na próbkowaniu i kwantowej diagonalizacji do przetwarzania próbek pobranych z zestawu 16-Qubitowych stanów bazy Kryłowa dla rosnących przedziałów czasowych. Stany te są realizowane na urządzeniu kwantowym przy użyciu troterizacji ewolucji czasowej. Aby uwzględnić wpływ szumów kwantowych, stosowana jest technika odtwarzania konfiguracji. Zakładając dobry stan początkowy i rzadkość stanu podstawowego, podejście to jest udowodnione jako efektywnie zbieżne.
Wzorzec można opisać w czterech krokach:
- Krok 1: Mapowanie na problem kwantowy
- Wygenerowanie zestawu stanów bazy Kryłowa (tj. obwodów troterizowanej ewolucji czasowej) dla rosnących przedziałów czasowych w celu estymacji stanu podstawowego
- Krok 2: Optymalizacja problemu
- Transpilacja obwodów dla Backend'u
- Krok 3: Wykonanie eksperymentów
- Pobranie próbek z obwodów przy użyciu prymitywu
Sampler
- Pobranie próbek z obwodów przy użyciu prymitywu
- Krok 4: Przetwarzanie wyników po stronie klasycznej
- Pętla samospójnego odtwarzania konfiguracji
- Przetwarzanie pełnego zestawu próbek bitstring, z wykorzystaniem wiedzy a priori o liczbie cząstek i średnim obsadzeniu orbitali obliczonym w ostatniej iteracji
- Probabilistyczne tworzenie partii podpróbek z odtworzonych bitstringów
- Projekcja i diagonalizacja fermionowego Hamiltonianu sieciowego w każdej próbkowanej podprzestrzeni
- Zapisanie minimalnej energii stanu podstawowego znalezionej we wszystkich partiach oraz aktualizacja średniego obsadzenia orbitali
- Pętla samospójnego odtwarzania konfiguracji
Krok 1: Mapowanie problemu na obwód kwantowy
Na początku utworzymy jednocząstkowe i dwucząstkowe Hamiltoniany opisujące jednowymiarowy model Andersona z pojedynczą domieszką (SIAM) z 7 miejscami kąpieli (8 elektronów w 8 orbitalach). Model ten służy do opisu magnetycznych domieszek wbudowanych w metale.
Następnie utworzymy 16-Qubitowe obwody Trottera używane do generowania kwantowej podprzestrzeni Kryłowa.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
n_bath = 7 # number of bath sites
V = 1 # hybridization amplitude
t = 1 # bath hopping amplitude
U = 10 # Impurity onsite repulsion
eps = -U / 2 # Chemical potential for the impurity
# Place the impurity on the first qubit
impurity_index = 0
# One body matrix elements in the "position" basis
h1e = -t * np.diag(np.ones(n_bath), k=1) - t * np.diag(np.ones(n_bath), k=-1)
h1e[impurity_index, impurity_index + 1] = -V
h1e[impurity_index + 1, impurity_index] = -V
h1e[impurity_index, impurity_index] = eps
# Two body matrix elements in the "position" basis
h2e = np.zeros((n_bath + 1, n_bath + 1, n_bath + 1, n_bath + 1))
h2e[impurity_index, impurity_index, impurity_index, impurity_index] = U
Następnie wygenerujemy kwantową podprzestrzeń Kryłowa przy użyciu zestawu troterizowanych obwodów kwantowych. Tutaj tworzymy funkcje pomocnicze do generowania stanu początkowego (referencyjnego) oraz ewolucji czasowej dla jednocząstkowej i dwucząstkowej części Hamiltonianu. Bardziej szczegółowy opis tego modelu i sposobu projektowania obwodów znajdziesz w artykule.
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
n_modes = n_bath + 1
nelec = (n_modes // 2, n_modes // 2)
dt = 0.2
Utar = scipy.linalg.expm(-1j * dt * h1e)
# The reference state
def initial_state(q_circuit, norb, nocc):
"""Prepare an initial state."""
for i in range(nocc):
q_circuit.append(XGate(), [i])
q_circuit.append(XGate(), [norb + i])
rot = XXPlusYYGate(np.pi / 2, -np.pi / 2)
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
q_circuit.append(rot, [j, j + 1])
q_circuit.append(rot, [norb + j, norb + j + 1])
q_circuit.append(rot, [j + 1, j + 2])
q_circuit.append(rot, [norb + j + 1, norb + j + 2])
# The one-body time evolution
free_fermion_evolution = ffsim.qiskit.OrbitalRotationJW(n_modes, Utar)
# The two-body time evolution
def append_diagonal_evolution(dt, U, impurity_qubit, num_orb, q_circuit):
"""Append two-body time evolution to a quantum circuit."""
if U != 0:
q_circuit.append(
CPhaseGate(-dt / 2 * U),
[impurity_qubit, impurity_qubit + num_orb],
)
Wygeneruj d stanów po ewolucji czasowej, które określają kwantową podprzestrzeń Kryłowa. Tutaj wybraliśmy d=8. Błąd wynikający z próbkowania stanów bazy Kryłowa zbiega się wraz ze wzrostem d. Zwróć uwagę, że specyfika tego problemu umożliwia efektywną kompilację ewolucji jednocząstkowej przy użyciu OrbitalRotationJW; jednak w ogólnym przypadku można użyć PauliEvolutionGate z Qiskit do implementacji troterizowanej ewolucji czasowej pełnego Hamiltonianu.
# Generate the initial state
qubits = QuantumRegister(2 * n_modes, name="q")
init_state = QuantumCircuit(qubits)
initial_state(init_state, n_modes, n_modes // 2)
init_state.draw("mpl", scale=0.4, fold=-1)
d = 8 # Number of Krylov basis states
circuits = []
for i in range(d):
circ = init_state.copy()
circuits.append(circ)
for _ in range(i):
append_diagonal_evolution(dt, U, impurity_index, n_modes, circ)
circ.append(free_fermion_evolution, qubits)
append_diagonal_evolution(dt, U, impurity_index, n_modes, circ)
circ.measure_all()
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
Krok 2: Optymalizacja problemu
Po utworzeniu troterizowanych obwodów zoptymalizujemy je dla docelowego sprzętu. Przed optymalizacją musimy wybrać urządzenie sprzętowe. Użyjemy fałszywego Backend'u 127-Qubitowego z qiskit_ibm_runtime do emulacji prawdziwego urządzenia. Aby uruchomić na prawdziwym QPU, wystarczy zastąpić fałszywy Backend prawdziwym. Więcej informacji znajdziesz w dokumentacji Qiskit IBM Runtime.
from qiskit_ibm_runtime.fake_provider.backends import FakeSherbrooke
backend = FakeSherbrooke()
Następnie skompilujemy obwody do docelowego Backend'u przy użyciu Qiskit.
from qiskit.transpiler import generate_preset_pass_manager
# The circuit needs to be transpiled to the AerSimulator target
pass_manager = generate_preset_pass_manager(optimization_level=3, backend=backend)
isa_circuits = pass_manager.run(circuits)
Krok 3: Wykonanie eksperymentów
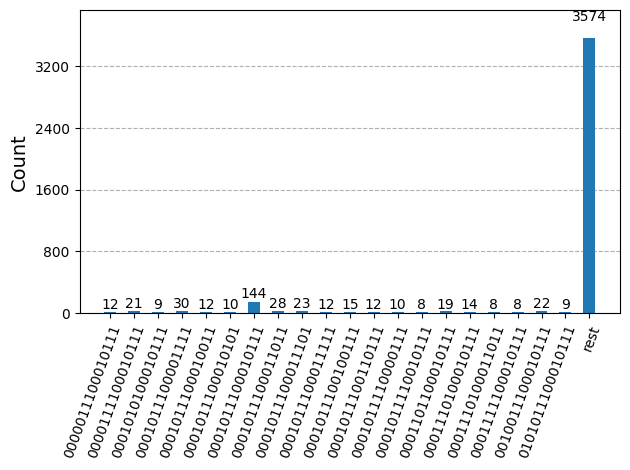
Po optymalizacji obwodów pod kątem wykonania na sprzęcie jesteśmy gotowi do uruchomienia ich na docelowym sprzęcie i zebrania próbek do estymacji energii stanu podstawowego. Tutaj używamy SamplerV2 z qiskit-ibm-runtime do symulacji zaszumionych próbek pobranych z Backend'u ibm_sherbrooke. Następnie łączymy zliczenia z każdego ze stanów bazy Kryłowa w jeden słownik zliczeń i wykreślamy 20 najczęściej próbkowanych bitstringów.
Uwaga: Qiskit Aer jest wymagany do symulacji próbek z skompilowanych obwodów.
from qiskit.primitives import BitArray
from qiskit.visualization import plot_histogram
from qiskit_ibm_runtime import SamplerV2 as Sampler
# Sample from the circuits
noisy_sampler = Sampler(backend, options={"simulator": {"seed_simulator": 24}})
job = noisy_sampler.run(isa_circuits, shots=500)
# Combine the counts from the individual Trotter circuits
bit_array = BitArray.concatenate_shots([result.data.meas for result in job.result()])
plot_histogram(bit_array.get_counts(), number_to_keep=20)
Krok 4: Przetwarzanie wyników po stronie klasycznej
Teraz uruchamiamy algorytm SQD przy użyciu funkcji diagonalize_fermionic_hamiltonian. Wyjaśnienia argumentów tej funkcji znajdziesz w dokumentacji API.
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e,
h2e,
bit_array,
samples_per_batch=300,
norb=n_modes,
nelec=nelec,
num_batches=3,
max_iterations=10,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.257128325607997
Subspace dimension: 3969
Subsample 1
Energy: -13.257128325607997
Subspace dimension: 3969
Subsample 2
Energy: -13.257128325607997
Subspace dimension: 3969
Iteration 2
Subsample 0
Energy: -13.319666127542039
Subspace dimension: 4096
Subsample 1
Energy: -13.420534292304595
Subspace dimension: 4624
Subsample 2
Energy: -9.136171594591085
Subspace dimension: 4624
Iteration 3
Subsample 0
Energy: -13.422491814612833
Subspace dimension: 4900
Subsample 1
Energy: -13.422491814612833
Subspace dimension: 4900
Subsample 2
Energy: -13.422491814612833
Subspace dimension: 4900
Iteration 4
Subsample 0
Energy: -13.422491814612833
Subspace dimension: 4900
Subsample 1
Energy: -13.422491814612833
Subspace dimension: 4900
Subsample 2
Energy: -13.422491814612833
Subspace dimension: 4900
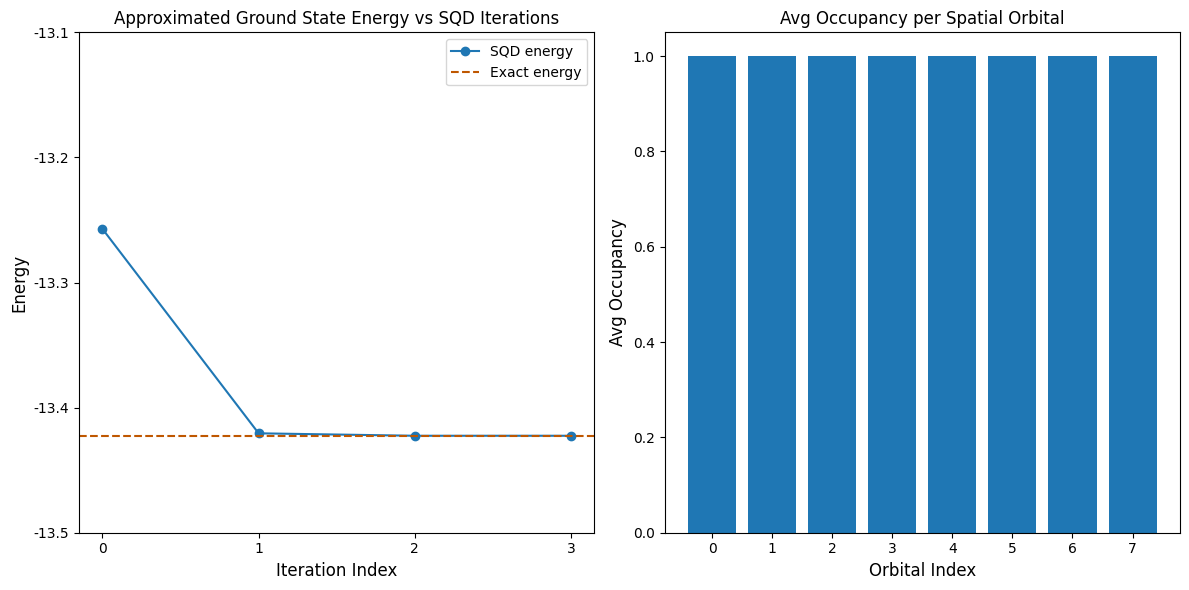
Teraz wykreślimy wyniki. Pierwszy wykres pokazuje, że po kilku iteracjach uzyskujemy dokładną energię stanu podstawowego.
Ten przykład jest na tyle mały, że możemy eksplorować pełną przestrzeń Hilberta, co widać w powyższych komunikatach. Pamiętaj, że pełna przestrzeń Hilberta ma wymiar (num_orbitals choose nelec_a) * (num_orbitals choose nelec_b). Dla tego problemu: (8 choose 4)**2 = 4900. Wymiary podprzestrzeni rosną dzięki ulepszonemu odtwarzaniu konfiguracji, a także dlatego, że algorytm SQD przenosi ważne konfiguracje z jednej iteracji do następnej. W ostatniej iteracji diagonalizujemy w całej przestrzeni Hilberta.
Drugi wykres pokazuje średnie obsadzenie każdego orbitalu przestrzennego we wszystkich rozwiązaniach partii. Widzimy, że z dużym prawdopodobieństwem każdy orbital zawiera jeden elektron.
import matplotlib.pyplot as plt
exact_energy = -13.422491814605827
min_es = [min(result, key=lambda res: res.energy).energy for result in result_history]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
yt1 = list(np.arange(-13.5, -13.1, 0.1))
ytl = [f"{i:.1f}" for i in yt1]
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="SQD energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(ytl)
axs[0].axhline(y=exact_energy, color="#BF5700", linestyle="--", label="Exact energy")
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - exact_energy):.5f}")
plt.tight_layout()
plt.show()
Exact energy: -13.42249
SQD energy: -13.42249
Absolute error: 0.00000