Poprawa estymacji energii hamiltonianów chemicznych za pomocą SQD
W tym samouczku implementujemy wzorzec Qiskit pokazujący, jak post-procesować zaszumione próbki kwantowe w celu znalezienia przybliżenia stanu podstawowego hamiltonianów chemicznych: cząsteczki w stanie równowagi w bazie 6-31G. Będziemy stosować podejście diagonalizacji kwantowej opartej na próbkach do przetwarzania próbek pobranych z ansatzu obwodu kwantowego na 36 Qubitach (w tym przypadku obwód LUCJ). Aby uwzględnić efekt szumów kwantowych, stosuje się technikę odtwarzania konfiguracji.
Wzorzec można opisać w czterech krokach:
- Krok 1: Odwzorowanie na problem kwantowy
- Generowanie ansatzu do estymacji stanu podstawowego
- Krok 2: Optymalizacja problemu
- Transpilacja ansatzu dla Backend
- Krok 3: Wykonanie eksperymentów
- Pobieranie próbek z ansatzu za pomocą prymitywu
Sampler
- Pobieranie próbek z ansatzu za pomocą prymitywu
- Krok 4: Post-processing wyników
- Pętla samo-spójnego odtwarzania konfiguracji
- Post-processing pełnego zestawu próbek bitstring, wykorzystując wcześniejszą wiedzę o liczbie cząstek oraz średnim obsadzeniu orbitali obliczonym w poprzedniej iteracji.
- Probabilistyczne tworzenie partii podpróbek z odtworzonych bitstringów.
- Rzutowanie i diagonalizacja molekularnego hamiltonianów na każdą próbkowaną podprzestrzeń.
- Zapisywanie minimalnej znalezionej energii stanu podstawowego we wszystkich partiach oraz aktualizacja średniego obsadzenia orbitali.
- Pętla samo-spójnego odtwarzania konfiguracji
W tym przykładzie hamiltonian elektronów oddziałujących ma ogólną postać:
/ to fermionowe operatory kreacji/anihilacji powiązane z -tym elementem bazy i spinem . i to jednociałowe i dwuciałowe całki elektronowe.
Przepływ pracy SQD z samo-spójnym odtwarzaniem konfiguracji przedstawiono na poniższym diagramie.
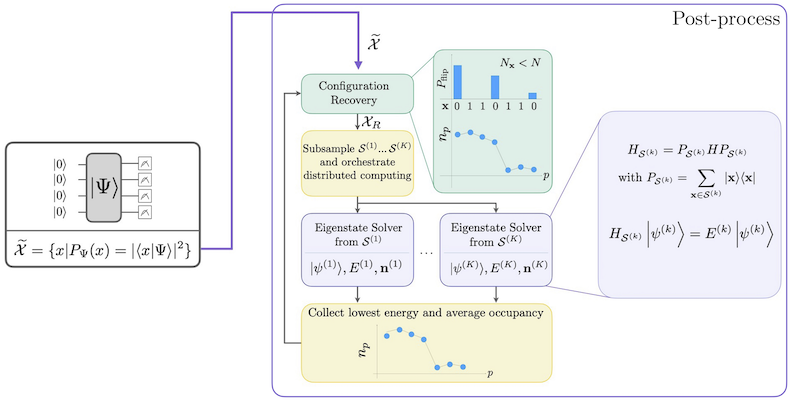
Wiadomo, że SQD działa dobrze, gdy docelowy stan własny jest rzadki: funkcja falowa jest podparta na zbiorze stanów bazowych , których rozmiar nie rośnie wykładniczo wraz z rozmiarem problemu. W tym scenariuszu diagonalizacja hamiltonianów rzutowanego na podprzestrzeń zdefiniowaną przez :
daje dobre przybliżenie docelowego stanu własnego. Rola urządzenia kwantowego polega na produkowaniu próbek należących wyłącznie do . Najpierw Circuit kwantowy przygotowuje stan na urządzeniu kwantowym. Stosowane jest kodowanie Jordana-Wignera. W konsekwencji elementy bazy obliczeniowej reprezentują stany Focka (konfiguracje/determinanty elektronowe). Circuit jest próbkowany w bazie obliczeniowej, dając zestaw zaszumionych konfiguracji . Konfiguracje są reprezentowane przez bitstring. Zestaw jest następnie przekazywany do klasycznego bloku post-processingu, gdzie stosuje się samo-spójną technikę odtwarzania konfiguracji. W ramach SQD rola urządzenia kwantowego polega na produkowaniu rozkładu prawdopodobieństwa.
Krok 1: Odwzorowanie problemu na obwód kwantowy
W tym samouczku będziemy przybliżać energię stanu podstawowego cząsteczki . Najpierw określimy cząsteczkę i jej właściwości. Następnie stworzymy ansatz lokalnego unitarnego klastra Jastrow (LUCJ) (Circuit kwantowy), aby generować próbki z komputera kwantowego do estymacji energii stanu podstawowego.
Najpierw określimy cząsteczkę i jej właściwości.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Następnie stworzymy ansatz. Ansatz LUCJ to parametryzowany Circuit kwantowy, który zainicjalizujemy amplitudami t2 i t1 otrzymanymi z obliczeń CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Użyjemy pakietu ffsim do stworzenia i zainicjalizowania ansatzu amplitudami t2 i t1 obliczonymi powyżej. Ponieważ nasza cząsteczka ma stan Hartree-Focka z zamkniętą powłoką, użyjemy zbalansowanej spinowo wariantu ansatzu UCJ — UCJOpSpinBalanced.
Ponieważ docelowy sprzęt IBM ma topologię heavy-hex, przyjmiemy wzorzec zig-zag dla interakcji Qubitów. W tym wzorcu orbitale (reprezentowane przez Qubity) o tym samym spinie połączone są topologią liniową (czerwone i niebieskie kółka), gdzie każda linia ma kształt zygzaka ze względu na łączność heavy-hex docelowego sprzętu. Również ze względu na topologię heavy-hex, orbitale różnych spinów mają połączenia co 4. orbital (0, 4, 8 itd.) (fioletowe kółka).
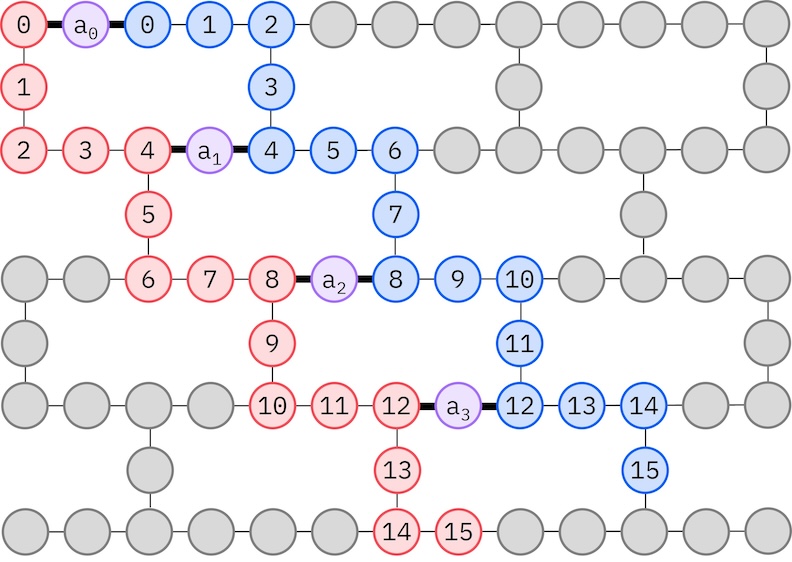
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Krok 2: Optymalizacja problemu
Następnie zoptymalizujemy nasz obwód dla docelowego sprzętu. Przed optymalizacją Circuit musimy wybrać urządzenie sprzętowe. Użyjemy fałszywego Backend z 127 Qubitami z qiskit_ibm_runtime, aby emulować rzeczywiste urządzenie. Aby uruchomić na prawdziwym QPU, wystarczy zastąpić fałszywy Backend prawdziwym. Więcej informacji znajdziesz w dokumentacji Qiskit IBM Runtime.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Następnie zalecamy wykonanie poniższych kroków w celu optymalizacji ansatzu i zapewnienia jego zgodności ze sprzętem.
- Wybierz fizyczne Qubity (
initial_layout) z docelowego sprzętu zgodnie z wzorcem zig-zag opisanym powyżej. Rozmieszczenie Qubitów w tym wzorcu prowadzi do wydajnego, zgodnego ze sprzętem Circuit z mniejszą liczbą Gate'ów. - Wygeneruj wieloetapowy menedżer przejść za pomocą funkcji generate_preset_pass_manager z Qiskit, podając wybrany
backendiinitial_layout. - Ustaw etap
pre_initswojego menedżera przejść naffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITzawiera przejścia Transpiler Qiskit, które dekompozytują Gate'y na rotacje orbitalne, a następnie scalają je, co skutkuje mniejszą liczbą Gate'ów w końcowym Circuit. - Uruchom menedżer przejść na swoim Circuit.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Krok 3: Wykonanie eksperymentów
Po zoptymalizowaniu Circuit do wykonania na sprzęcie jesteśmy gotowi uruchomić go na docelowym sprzęcie i zebrać próbki do estymacji energii stanu podstawowego. Ponieważ mamy tylko jeden obwód, użyjemy trybu wykonywania zadań Qiskit Runtime i wykonamy nasz obwód.
Uwaga: Zakomentowaliśmy kod do uruchomienia Circuit na QPU i pozostawiliśmy go jako odniesienie dla użytkownika. Zamiast uruchamiać na prawdziwym sprzęcie w tym przewodniku, po prostu wygenerujemy losowe próbki z rozkładu jednostajnego.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Krok 4: Post-processing wyników
Teraz uruchamiamy algorytm SQD za pomocą funkcji diagonalize_fermionic_hamiltonian. Wyjaśnienia argumentów tej funkcji znajdziesz w dokumentacji API.
Solver zawarty w dodatku SQD używa implementacji selected CI z PySCF, konkretnie pyscf.fci.selected_ci.kernel_fixed_space. Poniższy przykład pokazuje również, jak przekazywać argumenty słów kluczowych do tej funkcji za pośrednictwem dołączonego solvera. Tutaj przekazujemy argument max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
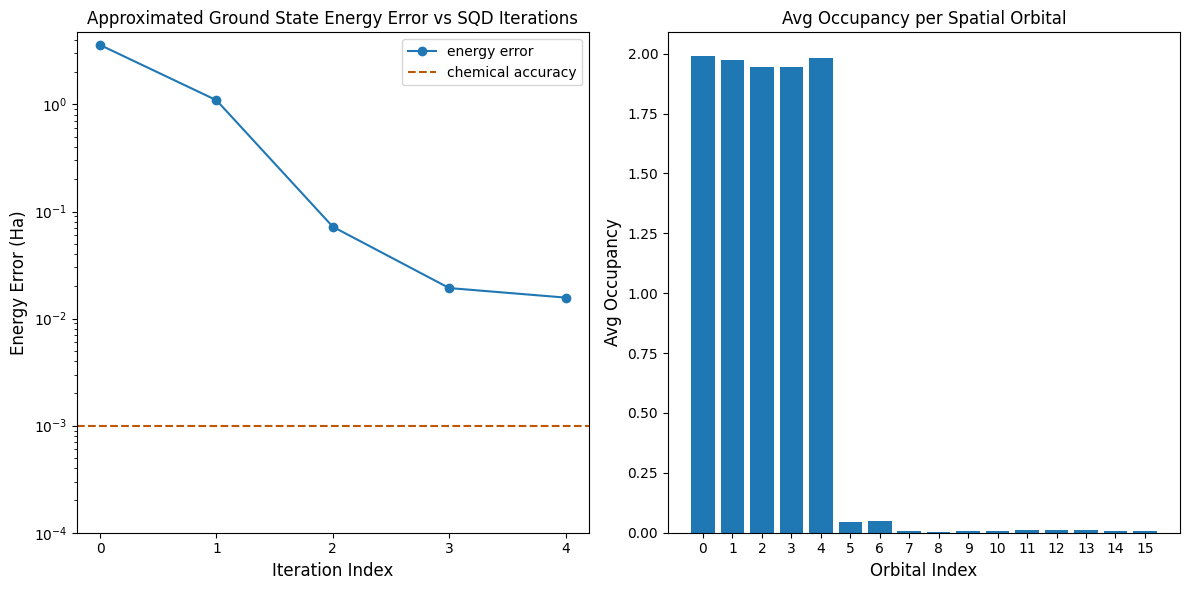
Teraz wykreślimy wyniki.
Pierwszy wykres pokazuje, że po kilku iteracjach szacujemy energię stanu podstawowego z dokładnością ~16 mH (dokładność chemiczna jest zazwyczaj przyjmowana jako 1 kcal/mol 1.6 mH). Pamiętaj, że próbki kwantowe w tej demonstracji były czystym szumem. Sygnał pochodzi tu z a priori wiedzy o strukturze elektronowej i molekularnym hamiltonianie.
Drugi wykres pokazuje średnie obsadzenie każdego orbitalu przestrzennego po ostatniej iteracji. Widać, że zarówno elektrony ze spinem w górę, jak i w dół z dużym prawdopodobieństwem obsadzają pierwsze pięć orbitali w naszych rozwiązaniach.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha