Mapowanie
Obejrzyj film o mapowaniu autorstwa Olivii Lanes lub otwórz film w osobnym oknie w serwisie YouTube.
Wprowadzenie
W tej lekcji skupimy się na pierwszym i często najtrudniejszym kroku w definiowaniu programu kwantowego: mapowaniu problemu, który ma być uruchomiony na komputerze kwantowym. Krok ten obejmuje sposób, w jaki użytkownik zaczyna od problemu obliczeniowego i przekłada go na coś, co można rozwiązać na komputerze kwantowym.
W lekcjach 2 i 3 tego kursu wspomnieliśmy, że etap mapowania jest pierwszym z czterech łącznie kroków w ramach wzorców Qiskit. Z tych lekcji możesz pamiętać, że celem mapowania jest przełożenie lub przepisanie problemu obliczeniowego na funkcję kosztu lub wartość oczekiwaną, którą możemy obliczyć za pomocą komputera kwantowego.
W lekcji 3 omówiliśmy jeden konkretny przykład z Max-Cut, obliczeniowo trudnym, ale bardzo powszechnym problemem w optymalizacji kombinatorycznej. W tamtym przykładzie przeszliśmy przez kilka kroków, aby przełożyć początkowy problem grafowy na taki, który można rozwiązać na komputerze kwantowym. Przekształciliśmy problem znajdowania maksymalnej liczby cięć w grafie na funkcję kosztu, przepisaliśmy tę funkcję kosztu jako Hamiltonian, a następnie przygotowaliśmy próbny stan kwantowy, którego stan podstawowy odpowiadał maksymalnemu cięciu. Na koniec skonstruowaliśmy obwód kwantowy reprezentujący interesujący nas próbny stan kwantowy, a następnie dodaliśmy konkretne bramki, aby umożliwić ewolucję stanu w czasie. Ta sekwencja kroków była całością w ramach mapowania. Chociaż dokładne kroki były unikalne dla problemu Max-Cut, tę samą ogólną procedurę można zastosować do wielu innych zastosowań, takich jak chemia kwantowa i symulacje kwantowe.
Mapowanie może być trudne. Nie istnieje uniwersalna strategia dla każdego pojedynczego problemu, więc może ono onieśmielać. W tej lekcji przyjrzymy się pewnym ogólnym zagadnieniom dotyczącym mapowania, a następnie zagłębimy się w kilka reprezentatywnych przykładowych problemów, aby zademonstrować różne sposoby mapowania problemu na komputer kwantowy.
Ogólne rozważania
Chociaż dokładna strategia, której używa się do mapowania problemu na komputer kwantowy, zależy od konkretnego problemu, ogólnie realizuje ona kilka głównych celów.
Po pierwsze, może być konieczne uproszczenie problemu, aby stał się wykonalny. Nie jest to cecha unikalna dla podejścia kwantowego – wszystkie dyscypliny naukowe używają uproszczonych modeli do badania interesujących je zjawisk, ignorując nieistotne szczegóły. W fizyce istnieje słynne wyrażenie, które doprowadza tę zasadę do skrajności: „załóżmy krowę sferyczną”. Często opisanie systemu dokładnie tak, jak się on prezentuje, jest zbyt trudne, ale zamiast tego możemy poczynić rozsądne uproszczenia, które nadal mogą prowadzić do pomocnych rozwiązań. Niektóre przykłady tego, jak możemy to zrobić w obliczeniach kwantowych, to ograniczenie rozmiaru lub głębokości obwodu poprzez wybór ansatzu efektywnego sprzętowo, skracanie złożonych ewolucji czasowych lub pomijanie członów Hamiltonianu, które w niewielkim stopniu przyczyniają się do końcowej energii stanu kwantowego.
Po drugie, mapowanie polega na zapisaniu problemu w taki sposób, aby komputer kwantowy mógł go zrozumieć. Często wiąże się to z zadaniem tych trzech pytań:
- Co nasze kubity będą reprezentować w naszym modelu?
- Czy nasz problem jest ciągły? Ponieważ komputery kwantowe są cyfrowe, jeśli problem jest ciągły, będziemy musieli znaleźć sposób na jego dyskretyzację.
- Wreszcie, czy topologia problemu jest zgodna z topologią sprzętu? Jeśli nie, być może będziemy musieli zastosować pewne sztuczki, aby to zadziałało.
Odnieśmy się do pierwszego pytania, które leży u podstaw wielu trudności ze zrozumieniem mapowania: Co może reprezentować kubit?
Kubity mogą być używane do reprezentowania wielu rzeczy. Pierwszą i być może najprostszą jest węzeł grafu. Grafy są używane do pokazania połączeń w wielu różnych rodzajach problemów matematycznych, a węzły są podstawowym elementem reprezentującym punkt lub podmiot w sieci. W zależności od tego, co reprezentuje cała sieć, może to być miasto, osoba lub ferromagnetyk w siatce.
Kubity mogą być również używane do reprezentowania bozonów i fermionów, chociaż zaznaczę tutaj, że jeden kubit nie jest dokładnie równy jednemu bozonowi czy jednemu fermionowi – jest to nieco bardziej skomplikowane, o czym omówimy dalej w lekcji.
Teraz otrzymujemy przykłady, które są nieco bardziej skomplikowane. W przypadku tych modeli nie ma już sensu mówić w kategoriach pojedynczych kubitów, zamiast tego potrzebujemy ich grup, aby utworzyć coś fizycznego. Na przykład grupa kubitów, tutaj reprezentowana na topologii ciężkiego heksagonu, może być używana do reprezentowania geometrycznych lokalizacji aminokwasów; łańcuchów polimerowych. Innym przykładem jest symulacja rozpraszania hadronów w modelach fizyki wysokich energii, którą można przeprowadzić, symulując ewolucję czasową hadronowego pakietu falowego. W tym przypadku rejestr kubitów może być używany do zakodowania różnych stanów pola kwantowego; stanu próżni tego pola lub pakietu falowego, który propaguje się na szczycie tej próżni.
Ale w tym momencie mówiliśmy wystarczająco abstrakcyjnie o stojącym przed nami wyzwaniu. Przyjrzyjmy się tym przykładom szczegółowo.
Przykłady mapowania
Max-Cut
Zacznijmy od pierwszego przykładu. Jednym z najbardziej bezpośrednich problemów mapowania jest ten, który omówiliśmy już dość dogłębnie: przykład Max-Cut. W tamtym problemie mapowanie było dla nas dość łatwe, ponieważ jeden kubit odpowiadał jednemu węzłowi w naszym grafie.
Przypomnijmy, że aby zmapować problem Max-Cut, wyraziliśmy funkcję kosztu jako Hamiltonian za pomocą sformułowania QUBO. Hamiltonowska funkcja kosztu to funkcja, która koduje optymalne rozwiązanie problemu w stanie podstawowym Hamiltonianu. Aby skonstruować Hamiltonian kosztu, użyliśmy klasy SparsePauliOp w Qiskit do określenia połączeń naszego grafu, a etap mapowania na operatory kwantowe został wykonany. A obwód kwantowy był po prostu ansatzem QAOA. Aby sobie przypomnieć, zobacz lekcję QAOA w skali użytkowej, w której omawiamy to wszystko znacznie bardziej szczegółowo.
W tamtej lekcji, nawet w 100-kubitowym przykładzie w skali użytkowej, połączenia grafu już pasowały do topologii naszego nadprzewodnikowego sprzętu. Nie musieliśmy więc zajmować się tym, jak radzić sobie z różnymi topologiami. Ale nie zawsze tak jest. Gdybyśmy mieli bardziej skomplikowany lub gęściej połączony graf niż przykłady, które do tej pory omawialiśmy, musielibyśmy zaimplementować serię bramek SWAP, aby zmodyfikować efektywne połączenia sprzętu. Jest to obsługiwane w drugim kroku wzorców Qiskit, transpilacji, ale należy o tym pamiętać nawet na etapie mapowania.
Zwijanie białek
Następnie zbadajmy przykład zamodelowany w artykule zatytułowanym "Resource-efficient quantum algorithm for protein folding", napisanym przez IBM® i współpracowników z University of New South Wales.
Trochę tła z biochemii: Białka są makrocząsteczkami złożonymi z długich łańcuchów aminokwasów. Łańcuchy te zwijają się w złożone struktury, które pełnią szeroką gamę funkcji biologicznych. Określenie struktury białka w trójwymiarowej przestrzeni oraz zrozumienie zależności między strukturą a funkcją należą do najtrudniejszych problemów współczesnej biochemii. Białka zwijają się w użyteczne struktury dzięki oddziaływaniom między aminokwasami. Gdy struktura skręca się i zwija, aminokwasy, które są daleko od siebie wzdłuż łańcucha, mogą znaleźć się tuż obok siebie i silnie oddziaływać.
Aby zamodelować to na komputerze kwantowym, potrzebujemy Hamiltonianu opisującego wszystkie te oddziaływania między aminokwasami. Następnie możemy przewidzieć końcową strukturę, znajdując stan, który zminimalizuje energię naszego Hamiltonianu. Tutaj skupimy się na tym, jak łańcuchy aminokwasów mogą być modelowane na komputerze kwantowym oraz jak możemy uzyskać odległości między aminokwasami do obliczenia energii oddziaływań. Dzięki temu zbierzemy wszystkie niezbędne wkłady do Hamiltonianu, które są niezbędne do symulacji go na komputerze kwantowym.
W rzeczywistych białkach aminokwasy mogą zajmować ciągłe spektrum możliwych lokalizacji. Jednak zastosujemy uproszczenie i ograniczymy je za pomocą modelu siatki, w którym każdy aminokwas zajmuje punkt na siatce. Tutaj autorzy użyli siatki tetraedrycznej. Krótka uwaga: tutaj kodujemy kierunek krawędzi, a nie same węzły, tak jak w problemie Max-Cut. Każdy kubit reprezentuje możliwą ścieżkę o jednym kroku wzdłuż siatki tetraedrycznej. Zauważ, że sąsiadujące miejsca zostały oznaczone jako A lub B z powodu ich różnych orientacji w siatce.
Łańcuch białka jest reprezentowany jako seria zakrętów lub kierunków na tej siatce. Każdy zakręt między aminokwasami może być w jednym z czterech kierunków, odpowiadających krawędziom czworościanu. Te cztery możliwe zakręty są kodowane za pomocą czterech kubitów w stanach 0001, 0010, 0100 lub 1000.
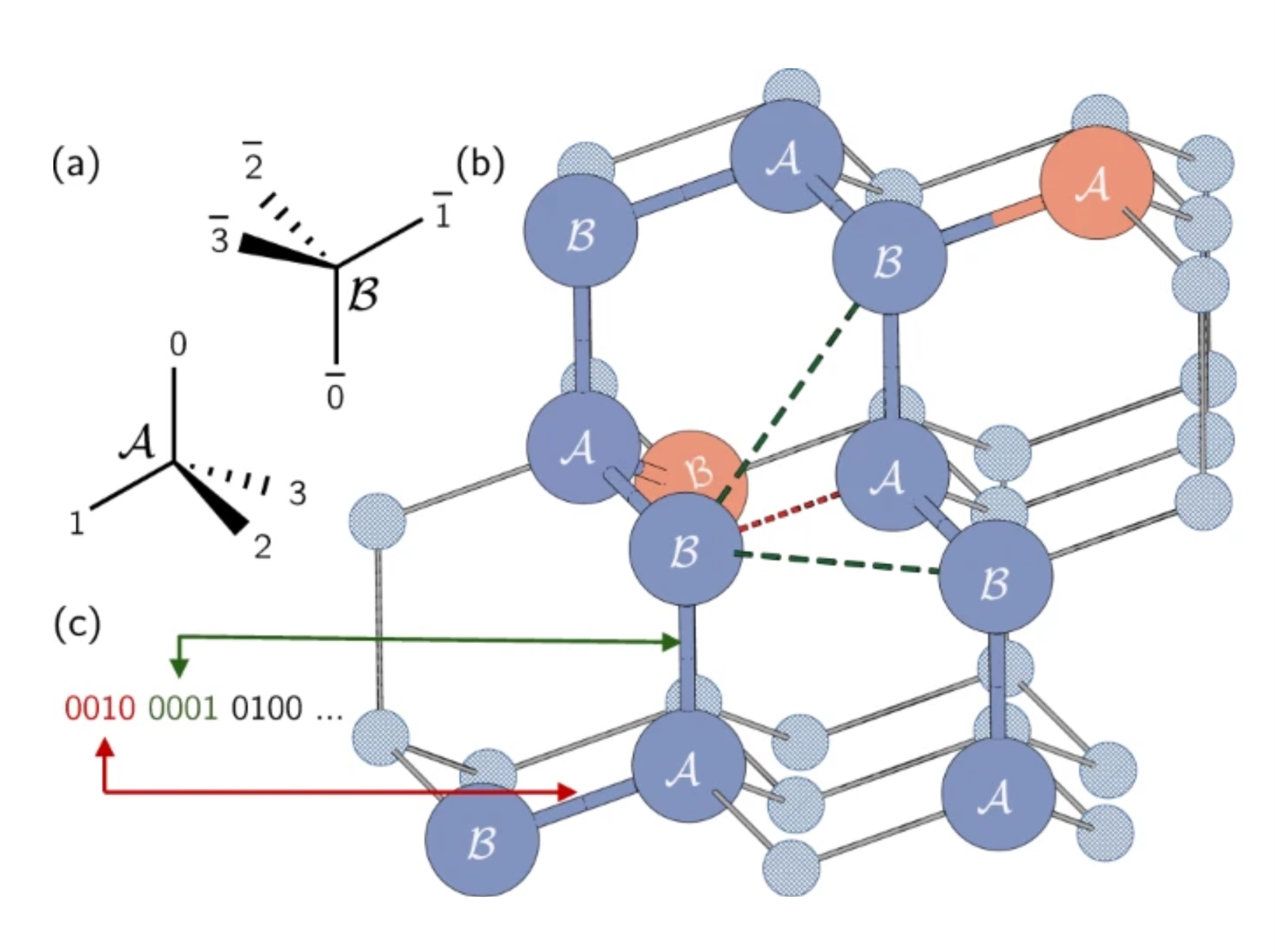
Spójrzmy na przykład na powyższym rysunku. Umieśćmy nasz pierwszy aminokwas w punkcie oznaczonym „B” zakreślonym na czerwono w naszej siatce tetraedrycznej. Kierunek od pierwszego aminokwasu do drugiego jest dowolny, ponieważ system zawsze można obrócić, aby ta krawędź wskazywała dowolny kierunek. Możemy więc umieścić nasz drugi aminokwas w punkcie pod pierwszym, oznaczonym „A”. Nie jest to tak łatwe do zauważenia, ale ścieżka od drugiego do trzeciego jest również dowolna. Wszystkie trzy wybory dałyby w efekcie dwie krawędzie o kącie około 109,5 stopnia między nimi. Wybór tej drugiej krawędzi po prostu determinuje orientację naszego białka w przestrzeni. Tak więc, bez utraty ogólności, możemy wybrać, aby pierwsze dwa zakręty były po prostu 0001 i 0010.
Dzięki tym uproszczeniom konfigurację łańcucha aminokwasów podaje to wyrażenie:
Do tej pory zmapowaliśmy krawędzie czworościanu na kubity, a nasz obwód kwantowy będzie ansatzem. Teraz musimy rozważyć, jak zakodować energię problemu w Hamiltonianie, tak aby jego stan podstawowy dawał nam optymalny wzór zwijania.
Dla dowolnej konfiguracji będzie istniała powiązana energia wynikająca z oddziaływań między aminokwasami. Oddziaływania te są najsilniejsze, gdy dwa aminokwasy znajdują się blisko siebie. Oczywiście aminokwasy, które sąsiadują w szkielecie łańcucha, zawsze będą oddziaływać ze sobą. Ale ponieważ białko może się skręcać i zwijać, inne pary aminokwasów również mogą oddziaływać. Na przykład aminokwasy 10 i 20 mogą znaleźć się w sąsiednich miejscach po zwinięciu białka. Potrzebujemy więc wzoru opisującego odległość między aminokwasami i przy użyciu informacji zakodowanych w kubitach konfiguracyjnych. W ten sposób możemy użyć ich odległości, aby określić, jak silnie oddziałują.
Najpierw wprowadźmy funkcję , która wskazuje, czy krawędź jest używana do zakrętu na aminokwasie . Tutaj może przyjąć dowolny z czterech możliwych kierunków. Na podstawie konfiguracji, od której zaczęliśmy powyżej, możemy zapisać te funkcje:
Następnie możemy zdefiniować różnicę w liczbie zakrętów oznaczonych jako na siatkach A i B od indeksu do indeksu jako :
Dlaczego mielibyśmy to robić? Otóż okazuje się, że obrót z węzła sieci A do B jest dokładnie znoszony przez obrót z węzła sieci B do A. Zatem aby poznać odległość aminokwasu w węźle od aminokwasu w węźle , wystarczy znaleźć różnicę między krokami wykonanymi wzdłuż krawędzi z węzłów A i węzłów B. Ponieważ węzły A i B muszą się naprzemiennie pojawiać wzdłuż szkieletu białka, jest to ujęte w czynniku . Ten sam argument odnosi się do wszystkich czterech typów krawędzi. Zatem całkowitą odległość między aminokwasami w krokach sieci tetraedrycznej można obliczyć za pomocą tego wyrażenia:
Ale jak uzyskać Hamiltonian z tego długiego równania opisującego całkowitą odległość między aminokwasami? Po pierwsze, możemy przeliczyć odległość w krokach sieci na przestrzeń euklidesową za pomocą prostej geometrii:
Następnie odległości te zostaną wykorzystane do obliczenia energii konfiguracji białka. W zależności od naszych celów możemy wyznaczyć odległość graniczną, poniżej której para jest uznawana za oddziałującą, lub możemy zrobić coś bardziej skomplikowanego.
Może nie jest to oczywiste, ale faktycznie zakończyliśmy już etap mapowania. Stany kubitów wskazują „obrót” białka w każdym węźle sieci, a zbiór obrotów wyznacza odległość między dowolną parą aminokwasów. Pary różnych rodzajów aminokwasów mają różne oddziaływania — niektóre przyciągające, inne odpychające. Jeśli używasz konfiguracji i odległości jedynie po to, by stwierdzić, czy znane oddziaływania między aminokwasami są „włączone” czy „wyłączone”, ich siły zostały już wyznaczone i można je po prostu odczytać z tabeli takiej jak ta:
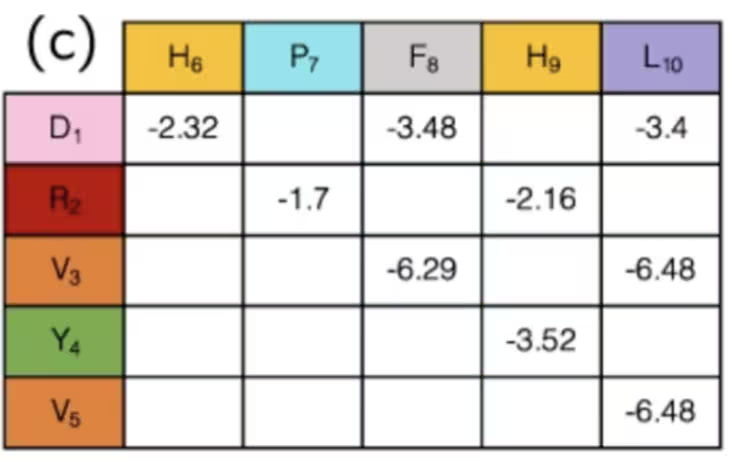
Podsumowując, w tym przykładzie kubity są używane do oznaczania kroków ścieżki wzdłuż sieci, które razem tworzą łańcuch aminokwasów. Symulując ich wyginanie i zwijanie, mamy nadzieję osiągać lepsze wyniki w badaniach medycznych. Pominęliśmy obliczanie kilku wyrazów tego Hamiltonianu, ponieważ były one bardzo specyficzne dla tego problemu, podczas gdy definiowanie kierunków w sieci można stosować bardziej ogólnie. Teraz, gdy masz ogólny Hamiltonian, zawsze będziesz chciał przetłumaczyć go na operatory Pauli, które są naturalne dla komputera kwantowego. O tym właśnie porozmawiamy dalej.
Transformacja Jordana-Wignera
Teraz zbadajmy, jak przetłumaczyć układ cząstek subatomowych na operatory Pauli.
Cząstki subatomowe dzielą się na dwie kategorie — bozony i fermiony. Bozony, takie jak fotony czy bozon Higgsa, podlegają pewnemu zestawowi reguł statystycznych. Fermiony, takie jak elektrony czy neutrina, podlegają innym. Kluczowa różnica między nimi polega na tym, że bozony mogą zajmować ten sam stan — nie ma ograniczenia liczby bozonów, które mogą znajdować się w stanie podstawowym lub w dowolnym stanie wzbudzonym. Fermiony natomiast są samolubne — wymagają, aby każda pojedyncza cząstka miała swój własny stan kwantowy.
Bozony mają również całkowite spiny, podczas gdy fermiony mają spin połówkowy, jak elektron o spinie 1/2, czy bardziej egzotyczne cząstki o spinie 3/2. Aby opisać układ cząstek, potrzebujemy opisu ich energii. Skupmy się na fermionach. Możemy zacząć od Hamiltonianu zapisanego w terminach tak zwanych operatorów c. Są to w zasadzie obiekty matematyczne, które odpowiadają tworzeniu lub anihilacji fermionu ze stanu w układzie. Zapisuje się je często jako i , gdzie to operator, który tworzy fermion w stanie , a to operator, który niszczy fermion w stanie .
Pamiętaj jednak, że komputery kwantowe zazwyczaj działają w bazie kubitowej z określonymi regułami reprezentowania układów fermionowych; nie działają one z natury w języku operatorów fermionowych. Aby wypełnić tę lukę, musimy odwzorować tę notację fermionową na operatory Pauli, które naturalnie odpowiadają bramkom kwantowym.
Istnieje kilka takich transformacji dla fermionów. Częstym wyborem jest transformacja Jordana-Wignera. Mapowania Bravyi-Kitaeva i parzystości to nowsze kodowania fermionowe. Operatory bozonowe można transformować za pomocą transformacji Holsteina-Primakoffa lub przez bezpośrednie odwzorowanie stanów Focka na podbazę dostępnych modów bozonowych, a także na inne sposoby. Aktywnie badane są również inne kodowania. Na razie skupimy się tylko na transformacie Jordana-Wignera.
Transformacja Jordana-Wignera polega na odwzorowaniu jednego fermionu na wiele kubitów. Dlaczego nie możemy po prostu przypisać jednego kubita do reprezentowania każdego elektronu? Ma to związek z nierozróżnialnością identycznych fermionów. Kubity są rozróżnialne, a elektrony nie. Na przykład z łatwością możemy etykietować i identyfikować poszczególne kubity na dowolnym urządzeniu. Ale nierozróżnialność elektronów oznacza, że w ogóle nie możemy ich etykietować. Musimy więc w rzeczywistości etykietować operatory zgodnie z zajętymi orbitalami, takimi jak 1s, 2p, 2p itd., zamiast konkretnych elektronów. Zatem każdy kubit pełni w przybliżeniu rolę orbitalu w cząsteczce, który jest albo zajęty, albo niezajęty. Ale to, jak to robimy, jest nieco bardziej skomplikowane. Mapowanie Jordana-Wignera śledzi antysymetrię i zapewnia poprawną statystykę całego układu fermionowego. Mapowanie Jordana-Wignera wyraża operatory fermionowe za pomocą operatorów Pauli, używając tych relacji:
Mapowanie Jordana-Wignera jest koncepcyjnie proste ze względu na jednoznaczną odpowiedniość między orbitalami a kubitami. Istnieją inne mapowania, które osiągają podobny cel, w tym mapowanie parzystości. Obliczenie parzystości stanu wymaga uwzględnienia wielu kubitów. W mapowaniu parzystości (i niektórych innych) interpretacja, że jeden kubit odpowiada jednemu orbitalowi, nie obowiązuje. Przejdźmy teraz przez prosty przykład. Załóżmy, że chcemy obliczyć jednokubitowe oddziaływanie . Zaczynamy od podstawienia naszych definicji operatorów kreacji i anihilacji.
Komutator . Zatem ostateczne wyrażenie przyjmuje postać:
Tak więc udało nam się przepisać wyrażenie fermionowe w terminach operatorów Pauli, które nasz komputer kwantowy będzie w stanie zrozumieć.
Omówmy szybko, jak zaimplementowalibyśmy mapowanie Jordana-Wignera w Qiskit. Ważne jest zrozumienie, jak działają tego rodzaju transformacje, ale niepraktyczne byłoby ręczne ich przeprowadzanie za każdym razem — zwłaszcza dla dużych układów. Na szczęście Qiskit ułatwia nam to dzięki funkcji SparsePauliOp.
Na wysokim poziomie kroki są następujące:
- Użyj funkcji
from_listz SparsePauliOp, aby utworzyć operator identycznościowy odpowiadający rozmiarowi mapowanej przestrzeni parametrów. - Zgodnie z definicją operatorów kreacji i anihilacji pokazaną wcześniej, użyj funkcji
from_listz SparsePauliOp do zdefiniowania operatorów Pauli , , . Spowoduje to odwzorowanie fermionowych operatorów kreacji i anihilacji na kubitowe operatory spinowe, kodując fermionową liczbę obsadzeń w bazie obliczeniowej kubitów. - Wygeneruj pożądany Hamiltonian w bazie Pauli, stosując powyższe operatory do interesujących nas orbitali. Zwykle odpowiada to utworzeniu macierzy jednostkowej, która reprezentuje rdzeniowe (nieoddziałujące) orbitale, a następnie zastosowaniu operatorów kreacji i anihilacji do przestrzeni aktywnej, z wagami odpowiadającymi szczegółom rozwiązywanego problemu.
Teraz, gdy w pełni rozumiemy schemat mapowania Jordana-Wignera, zobaczmy bardziej skomplikowany przykład. Być może pamiętasz artykuł zatytułowany „Scalable Circuits for Preparing Ground States on Digital Quantum Computers: The Schwinger Model Vacuum on 100 Qubits” z poprzedniej lekcji. Nie będziemy ponownie omawiać tego artykułu szczegółowo — skupimy się tylko na mapowaniu Jordana-Wignera, które jest używane do wyrażenia Hamiltonianu węzłów sieci o węzłach dla modelu Schwingera przy braku pola elektrycznego.
Tutaj znacznie trudniej jest precyzyjnie wskazać, co reprezentuje jeden kubit w tym modelu, ponieważ tylko zbiór kubitów razem tworzy coś fizycznego — w tym przypadku paczkę falową. Zamiast tego można z grubsza myśleć o kubitach jako o zdyskretyzowanych fragmentach przestrzeni. Tutaj jest objętością sieci, w której każdy element (komórka elementarna) składa się z dwóch kubitów. Operatory fermionowe, które widzieliśmy na poprzednim slajdzie, opisują amplitudę funkcji falowej w konkretnym węźle. Zatem nasz Hamiltonian zawiera te fermionowe operatory kreacji i anihilacji. Używamy więc transformacji Jordana-Wignera do odwzorowania tych operatorów na operatory Pauli.
gdzie jest operatorem Pauli , a jest Gdy już mamy Hamiltonian zapisany w tym formacie, trudna część etapu mapowania dobiegła końca i można go teraz łatwo zapisać jako obwód w operatorach Pauli.
Podsumowanie
Omówiliśmy cztery przykłady tego, jak konkretne techniki mapowania były ostatnio stosowane w dziedzinie obliczeń kwantowych, zaczynając od najprostszych i przechodząc do zastosowania transformacji Jordana-Wignera w dynamice hadronów. Wiele z tego materiału jest bardzo techniczne i jeśli nie spotkałeś się z nim wcześniej, może wydawać się bardzo onieśmielające. Jednak z czasem staje się łatwiejsze — właśnie dlatego ten kurs nosi nazwę Quantum Computing in Practice. Nie jest to coś, co ktokolwiek może po prostu podchwycić i od razu stosować — wymaga nauki, zastanowienia i zapewne chwil frustracji. Zachęcam jednak do zaakceptowania tego dyskomfortu i naprawdę zgłębiania pytań, które będą pojawiać się w trakcie dalszej nauki. To jedyna droga do opanowania tej wiedzy.