Wariacyjny Kwantowy Solver Własny (VQE)
W tym module studenci muszą mieć skonfigurowane środowisko Python oraz zainstalowane najnowsze wersje następujących pakietów:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Aby skonfigurować i zainstalować te pakiety, zapoznaj się z przewodnikiem Instalacja Qiskit. Aby uruchamiać zadania na prawdziwych komputerach kwantowych, studenci muszą założyć konto IBM Cloud, postępując zgodnie z krokami opisanymi w przewodniku Konfiguracja konta IBM Cloud.
Moduł ten był testowany i wykorzystał łącznie około 8 minut czasu QPU. Jest to szacunkowa wartość — rzeczywiste zużycie może się różnić.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Wprowadzenie
Od czasu opracowania kwantowomechanicznego modelu na początku XX wieku naukowcy rozumieją, że elektrony nie poruszają się po stałych orbitach wokół jądra atomu, lecz istnieją w obszarach prawdopodobieństwa zwanych orbitalami. Orbitale te odpowiadają konkretnym, dyskretnym poziomom energii, które elektrony mogą zajmować. Elektrony naturalnie przebywają na najniższych dostępnych poziomach energii, znanych jako stan podstawowy. Jeśli jednak elektron pochłonie wystarczającą ilość energii, może przeskoczyć na wyższy poziom energii, przechodząc w stan wzbudzony. Stan wzbudzony jest przejściowy — elektron ostatecznie powraca na niższy poziom energii, uwalniając pochłoniętą energię, często w postaci światła. Ten fundamentalny proces pochłaniania i emitowania energii jest kluczowy dla zrozumienia, jak atomy oddziałują ze sobą i tworzą wiązania.
Gdy atomy łączą się, tworząc cząsteczki, ich orbitale atomowe łączą się, tworząc orbitale molekularne. Rozmieszczenie i poziomy energii elektronów w tych orbitalach molekularnych decydują o właściwościach powstałej cząsteczki oraz o sile wiązań chemicznych. Na przykład, podczas tworzenia cząsteczki wodoru () z dwóch pojedynczych atomów wodoru, elektron z każdego atomu zajmuje orbitale atomowe. Gdy atomy zbliżają się do siebie, orbitale atomowe nakładają się i łączą, tworząc nowe orbitale molekularne — jeden o niższej energii (orbital wiążący) i jeden o wyższej energii (orbital antywiążący). Dwa elektrony, po jednym z każdego atomu wodoru, będą preferować zajęcie orbitalu wiążącego o niższej energii, co prowadzi do powstania stabilnego wiązania kowalencyjnego utrzymującego cząsteczkę . Różnica energii między oddzielonymi atomami a utworzoną cząsteczką, a w szczególności energia elektronów w orbitalach molekularnych, determinuje stabilność i właściwości wiązania.
W kolejnych sekcjach zbadamy ten proces tworzenia cząsteczek, skupiając się na cząsteczce . Użyjemy prawdziwego komputera kwantowego w połączeniu z klasycznymi technikami optymalizacji, aby wyznaczyć energię tego prostego, lecz fundamentalnego procesu. Eksperyment ten stanowi praktyczną demonstrację tego, jak obliczenia kwantowe można zastosować do rozwiązywania problemów z zakresu chemii obliczeniowej, dostarczając wglądu w rolę energii elektronów.
VQE — wariacyjny algorytm kwantowy dla problemów wartości własnych
Techniki przybliżeń w chemii — zasada wariacyjna i zestaw bazowy
Wkład Erwina Schrödingera w mechanikę kwantową nie ogranicza się do wprowadzenia nowego modelu elektronowego — fundamentalnie ustanowił on mechanikę falową, rozwijając słynne zależne od czasu równanie Schrödingera:
Tutaj jest operatorem Hamiltona, który reprezentuje całkowitą energię układu, a jest funkcją falową zawierającą wszystkie informacje o stanie kwantowym układu. (Uwaga: jest całkowitą pochodną po czasie i nie uwzględniamy tu jawnie wartości własnej energii .)
Jednak w wielu praktycznych zastosowaniach — takich jak wyznaczanie dozwolonych poziomów energii atomów i cząsteczek — korzystamy zamiast tego z niezależnego od czasu równania Schrödingera (równania na wartości własne energii), które jest wyprowadzane z postaci zależnej od czasu przy założeniu stanu stacjonarnego. Stan stacjonarny to stan kwantowy, w którym gęstość prawdopodobieństwa znalezienia cząstki w danym punkcie przestrzeni nie zmienia się w czasie.
W tej postaci reprezentuje wartość własną energii odpowiadającą stanowi kwantowemu . Hamiltonian uwzględnia różne wkłady energetyczne, takie jak energia kinetyczna elektronów i jąder, siły przyciągania między elektronami a jądrami oraz siły odpychania między elektronami.
Rozwiązanie równania na wartości własne energii pozwala nam obliczyć skwantowane poziomy energii układów atomowych i molekularnych. Jednak dla cząsteczek dokładne jego rozwiązanie jest trudne, ponieważ funkcja falowa , opisująca przestrzenny rozkład elektronów, jest złożona i wielowymiarowa.
W związku z tym naukowcy stosują techniki przybliżeń, aby uzyskać praktyczne i dokładne rozwiązania. W tym module skupimy się na dwóch kluczowych metodach:
-
Zasada wariacyjna
Ta metoda aproksymuje funkcję falową i dostosowuje ją tak, aby jak najbardziej zbliżyć się do docelowej energii, zazwyczaj energii stanu podstawowego układu. Kluczowa idea zasady wariacyjnej jest prosta:
- Jeśli zgadniemy funkcję falową (tzw. „funkcję próbną"), obliczona z niej energia będzie zawsze równa lub wyższa niż energia stanu podstawowego () układu.
- Dostosowując parametry w funkcji próbnej, , możemy uzyskiwać coraz lepsze przybliżenie energii stanu podstawowego.
- Dokładność tej metody silnie zależy od wyboru funkcji próbnej . Źle dobrana funkcja próbna może prowadzić do oszacowania energii dalekiego od rzeczywistości.
-
Przybliżenie zestawem bazowym
Druga metoda przybliżeń dotyczy etapu konstruowania funkcji falowej — podejście zestawu bazowego. W chemii kwantowej dokładne rozwiązanie równania Schrödingera dla cząsteczek jest praktycznie niemożliwe. Zamiast tego aproksymujemy złożoną, wieloelektronową funkcję falową, budując ją z prostszych, z góry zdefiniowanych funkcji matematycznych. Zestaw bazowy to zbiór takich znanych funkcji matematycznych, zazwyczaj skupionych na atomach w cząsteczce, które służą jako elementy składowe do reprezentowania kształtu i zachowania elektronów w układzie. Można to porównać do próby odtworzenia szczegółowej rzeźby z użyciem wyłącznie standardowych klocków LEGO — im więcej rodzajów i rozmiarów klocków masz (im większy zestaw bazowy), tym dokładniej możesz przybliżyć oryginalny kształt.
Funkcje bazowe są często inspirowane analitycznymi rozwiązaniami dla prostych układów, jak atom wodoru, i przyjmują postaci funkcji gaussowskich lub typu Slatera, choć nadal są przybliżeniami. Zamiast pracować z teoretycznie „dokładnymi", lecz nieobliczalnymi pełnymi orbitalami molekularnymi, wyrażamy je jako kombinację liniową (sumę ze współczynnikami) tych funkcji bazowych. Metoda ta jest znana jako podejście Liniowej Kombinacji Orbitali Atomowych (LCAO), gdy funkcje bazowe przypominają orbitale atomowe. Poprzez optymalizację współczynników tej kombinacji liniowej możemy znaleźć najlepszą możliwą przybliżoną funkcję falową i energię w granicach wybranego zestawu bazowego.
- Im więcej funkcji zawiera zestaw bazowy, tym lepsze przybliżenie, ale kosztem większego nakładu obliczeniowego.
- Mały zestaw bazowy daje przybliżone szacunki, podczas gdy duży zestaw bazowy zapewnia dokładniejsze wyniki kosztem większych zasobów obliczeniowych.
Podsumowując: aby uczynić obliczenia wykonalnymi i zredukować koszt obliczeniowy, stosujemy zasadę wariacyjną poprzez aproksymację funkcji falowej, co zmniejsza złożoność obliczeniową i umożliwia iteracyjną optymalizację minimalizującą energię. Podejście zestawem bazowym upraszcza natomiast obliczenia, reprezentując orbitale atomowe jako kombinację z góry zdefiniowanych funkcji, zamiast bezpośrednio wyznaczać ciągłą funkcję falową.
Sprawdź swoją wiedzę
Rozważ próbną funkcję falową , gdzie jest stałą normalizacyjną, a jest regulowanym parametrem.
(a) Znormalizuj próbną funkcję falową, wyznaczając tak, aby
.
(b) Oblicz wartość oczekiwaną Hamiltonianu danego wzorem:
gdzie , co odpowiada potencjałowi prostego oscylatora harmonicznego.
(c) Użyj zasady wariacyjnej, aby znaleźć optymalne , minimalizując
Odpowiedź:
(a) Aby znormalizować daną próbną funkcję falową:
Skorzystaj z całki gaussowskiej:
podstawiając , otrzymujemy:
(b) Hamiltonian oscylatora harmonicznego to:
- Wartość oczekiwana energii kinetycznej
Obliczając drugą pochodną:
Zatem:
Korzystając ze standardowych wyników całek gaussowskich:
- Wartość oczekiwana energii potencjalnej
Korzystając z:
otrzymujemy:
- Całkowita wartość oczekiwana energii
(c) Wyznacz optymalne minimalizujące energię
Różniczkując:
Rozwiązując:
Podstawiając do :
co jest zgodne z dokładną energią stanu podstawowego kwantowego oscylatora harmonicznego.
VQE (Variational Quantum Eigensolver)
Variational quantum eigensolver (VQE) to główna metoda, którą wykorzystamy do zbadania procesu . Przyjrzyjmy się teraz temu, czym jest VQE i jak działa. Najpierw jednak zatrzymajmy się na chwilę i przeanalizujemy jedną bardzo ważną kwestię w poniższym pytaniu sprawdzającym.
Sprawdź swoją wiedzę
Skoro mamy już tyle strategii do rozwiązywania problemów z chemii, to po co nam komputer kwantowy? I jaki jest cel jednoczesnego korzystania z komputerów kwantowych i klasycznych?
Odpowiedź:
Obliczenia kwantowe mają szansę zrewolucjonizować chemię, rozwiązując problemy, z którymi klasyczne komputery sobie nie radzą ze względu na wykładnicze skalowanie stanów kwantowych. Richard Feynman słynnie zauważył, że aby symulować przyrodę, obliczenia muszą być również kwantowe [ref 1].
Przykładowo, symulacja kofeiny przy użyciu najprostszego zbioru bazowego (STO-3G) wymagałaby bitów — znacznie więcej niż łączna liczba gwiazd w obserwowalnym wszechświecie () [ref 2]. Komputer kwantowy może opisać orbitale elektronowe kofeiny za pomocą 160 qubitów.
Komputery kwantowe w naturalny sposób przetwarzają kwantowe oddziaływania przy użyciu superpozycji i splątania, co stanowi obiecującą drogę do dokładnych symulacji molekularnych. Co więcej, możemy łączyć zalety komputerów kwantowych (symulacja elektronów) z zaletami komputerów klasycznych (wstępne i końcowe przetwarzanie danych, zarządzanie procesem algorytmu, optymalizacja itd.). Oczekuje się, że przyspieszy to odkrywanie nowych materiałów, projektowanie leków oraz przewidywanie przebiegu reakcji, ograniczając kosztowne eksperymenty metodą prób i błędów. [ref 3][ref 4]
Jeśli chcesz dowiedzieć się, dlaczego komputery kwantowe są potrzebne do rozwiązywania problemów chemicznych i dlaczego warto korzystać jednocześnie z zasobów obliczeniowych kwantowych i klasycznych, zapoznaj się z poniższymi artykułami:
Wróćmy teraz do VQE.
VQE łączy moc komputerów kwantowych z komputerami klasycznymi, stosując w podstawowym ujęciu zasadę wariacyjną do wyznaczenia energii stanu podstawowego układu. Aby zrozumieć VQE, rozbij go na trzy części:
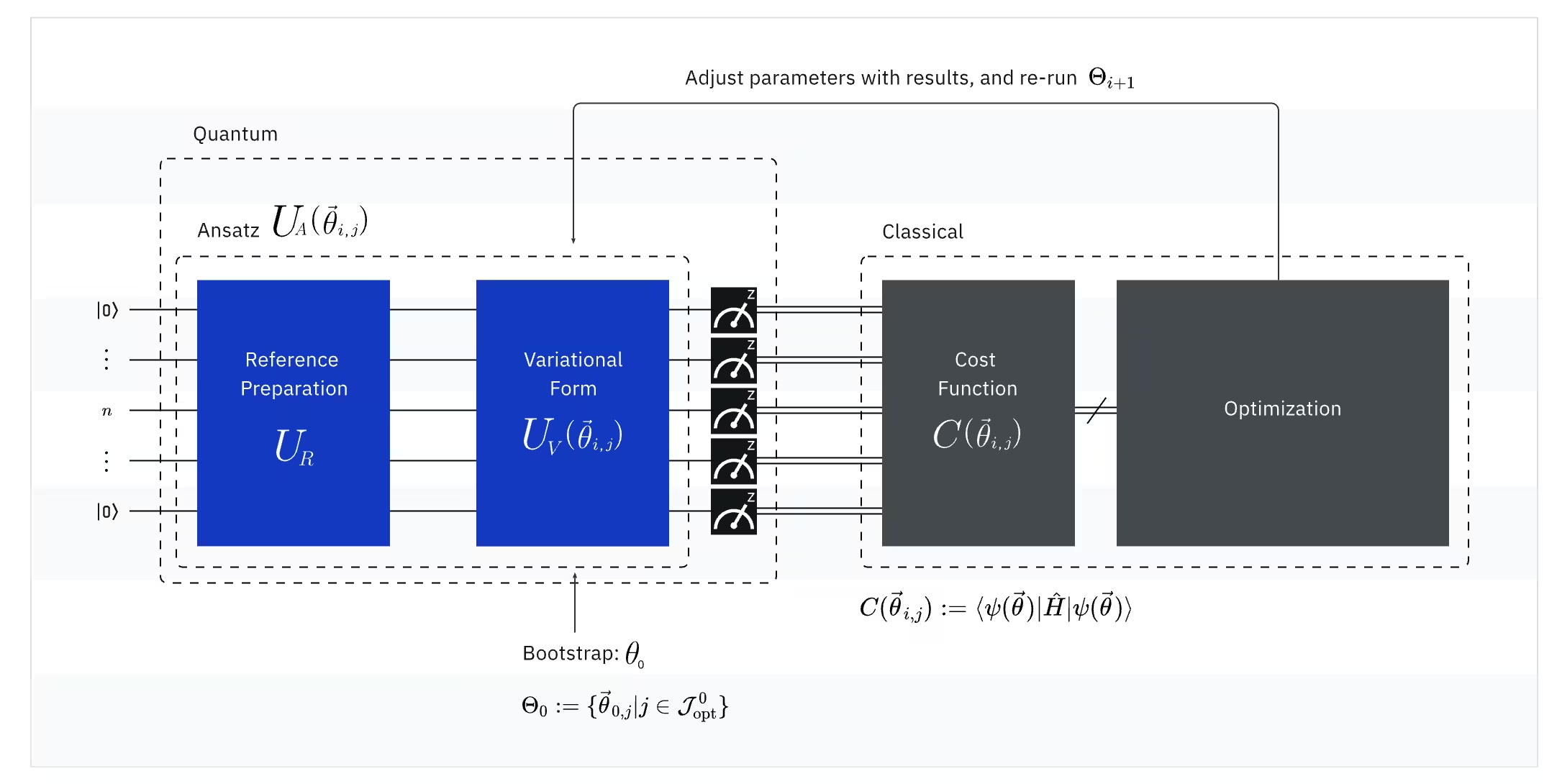
(Kwantowy) Obserwowalny: Hamiltonjan molekularny (energia cząsteczki)
W VQE molekularny/atomowy Hamiltonjan jest obserwowalny, co oznacza, że możemy zmierzyć jego wartość w eksperymencie. Naszym celem jest znalezienie jak najniższej energii (energii stanu podstawowego) cząsteczki. W tym celu używamy próbnego stanu kwantowego, wygenerowanego przez sparametryzowany Circuit kwantowy (ansatz). Mierzymy obserwowalny i optymalizujemy stan kwantowy, dopóki nie osiągniemy jak najniższej energii.
Zbiór bazowy użyty dla Hamiltonianu molekularnego określa liczbę potrzebnych qubitów i bezpośrednio wpływa na dokładność VQE. Wybór odpowiedniego zbioru bazowego ma kluczowe znaczenie dla zachowania równowagi między wydajnością a precyzją. Aby uprościć obliczenia bez zmiany zbioru bazowego, możemy stosować strategie takie jak narzucanie symetrii i redukcja przestrzeni aktywnej. Wiele cząsteczek ma symetryczne kształty (jak motyl lub śnieżynka), co oznacza, że niektóre ich fragmenty zachowują się identycznie. Zamiast obliczać wszystko oddzielnie, możemy skupić się wyłącznie na unikalnych częściach, oszczędzając zasoby kwantowe — czyli wykorzystując symetrię. W redukcji przestrzeni aktywnej bierzemy pod uwagę jedynie ważne orbitale, ponieważ nie wszystkie elektrony mają istotny wpływ na energię molekularną. Elektrony blisko jądra pozostają w dużej mierze niezmienione, podczas gdy inne wpływają na wiązania. Stosując te metody, możemy zwiększyć efektywność VQE przy zachowaniu dokładności.
Po uzyskaniu Hamiltonianu molekularnego przy użyciu odpowiedniego zbioru bazowego i opisanych strategii, musimy przekształcić ten Hamiltonjan do postaci odpowiedniej dla komputerów kwantowych. Odwzorowanie problemów na operatory Pauliego może być dość skomplikowane. Jest to szczególnie prawdziwe w chemii kwantowej, która operuje na nierozróżnialnych cząstkach (elektronach), podczas gdy qubity są rozróżnialne. Nie będziemy tu wchodzić w szczegóły tych odwzorowań, lecz odsyłamy do poniższych zasobów. Ogólne omówienie odwzorowania problemu na operatory kwantowe znajdziesz w kursie Quantum computing in practice. Bardziej szczegółowe omówienie odwzorowania problemów chemicznych na operatory kwantowe znajdziesz w kursie Quantum chemistry with VQE.
W tym module dostarczymy ci odpowiednie (jednoQubitowe) Hamiltoniany dla i , abyśmy mogli skupić się na korzystaniu z komputera kwantowego. Te jednoQubitowe Hamiltoniany są przygotowane przy użyciu zbioru bazowego STO-6G i odwzorowania Jordana-Wignera, które jest najprostszym odwzorowaniem o najłatwiejszej do zrozumienia interpretacji fizycznej — mapuje ono obsadzenie jednego spinorbitalu na obsadzenie jednego qubitu. Zastosowaliśmy również technikę redukcji qubitów opartą na symetrii Hamiltonianu, która wykorzystuje wzorce zachowania obsadzeń spinowych do zmniejszenia liczby qubitów. Dla cząsteczki zakładamy, że odległość między dwoma atomami wodoru wynosi 0.735 .
(Kwantowy) Ansatz: Próbna funkcja falowa (jak zbudować próbny stan kwantowy za pomocą Circuit kwantowego)
W VQE ansatz (l.mn.: ansätze) składa się z dwóch kluczowych elementów. Pierwszym jest przygotowanie stanu początkowego, które ustawia stan qubitu przez zastosowanie Gate kwantowych bez parametrów wariacyjnych. Drugim elementem jest sparametryzowany Circuit kwantowy — specjalny Circuit kwantowy z regulowanymi parametrami, podobnymi do pokręteł w radiu. Parametry te będą wykorzystywane przez ostatnią część — klasyczny optymalizator — aby pomóc nam osiągnąć jak najlepszy stan podstawowy.
W sekcji dotyczącej zasady wariacyjnej dowiedzieliśmy się, że jakość próbnego stanu wpływa na jakość wyników algorytmu wariacyjnego. Oznacza to, że wybór dobrego ansatzu jest ważny w VQE. Jest to kolejny bogaty i złożony temat. Nie będziemy tu omawiać różnych typów ansatzu ani ich pochodzenia. Jeśli chcesz dowiedzieć się więcej o sparametryzowanych Circuit kwantowych i ansatzu, możesz zapoznać się z lekcją Ansatz and variational form z kursu Variational algorithm design, który zawiera szczegółowe wyjaśnienia i przykłady ansätze.
Ponieważ w tym module będziemy używać jednoQubitowego Hamiltonianu, potrzebujemy jednoQubitowego sparametryzowanego Circuit kwantowego jako ansatzu. W następnej sekcji zobaczymy trzy typy jednoQubitowych ansätze. Porównamy je i omówimy kluczowe kwestie przy wyborze ansatzu.
(Klasyczny) Optymalizator: dostrajanie Circuit kwantowego
Po tym jak komputer kwantowy zmierzy energię obserwowalnego z ansatzu, parametry ansatzu i wartość energii są wysyłane do klasycznego optymalizatora w celu dostrojenia. Ten proces optymalizacji jest wykonywany na komputerze klasycznym, zazwyczaj z użyciem ogólnych pakietów naukowych, takich jak SciPy.
Klasyczny optymalizator traktuje zmierzoną energię jako funkcję kosztu. W problemach optymalizacyjnych funkcja kosztu (zwana też funkcją celu) to funkcja matematyczna mierząca, jak „dobra" jest dane rozwiązanie. Celem optymalizatora jest znalezienie zestawu parametrów minimalizujących tę funkcję kosztu. W kontekście wyznaczania energii stanu podstawowego cząsteczki sama energia pełni rolę funkcji kosztu — chcemy znaleźć parametry naszego Circuit kwantowego (nasze „rozwiązanie"), które dają jak najniższą energię. Klasyczny optymalizator używa zmierzonej wartości energii (kosztu) i wyznacza następny zestaw zoptymalizowanych parametrów dla kwantowego ansatzu. Zaktualizowane parametry są następnie wysyłane z powrotem do obwód kwantowego i proces się powtarza. Przy każdej iteracji klasyczny optymalizator dostosowuje parametry, starając się obniżyć energię (zminimalizować funkcję kosztu), aż do spełnienia określonego kryterium zbieżności — co w założeniu zapewnia znalezienie jak najniższej energii (odpowiadającej stanowi podstawowemu cząsteczki dla danej odległości wiązania i zbioru bazowego).
Pakiety naukowe, takie jak SciPy, oferują wiele strategii optymalizacji. Więcej na ten temat znajdziesz w lekcji Optimization loops kursu Variational algorithm design. Tutaj użyjemy COBYLA (Constrained Optimization BY Linear Approximations) — algorytmu optymalizacji odpowiedniego do skomplikowanych przestrzeni energetycznych. COBYLA w szczególności nie próbuje obliczać gradientu badanej funkcji; jest to tzw. optymalizator bezgradientowy. Wyobraź sobie, że próbujesz znaleźć najwyższy szczyt w paśmie górskim z zamkniętymi oczami. Ponieważ nie widzisz całego krajobrazu, robisz małe kroki w różnych kierunkach, sprawdzając, czy idziesz w górę czy w dół. COBYLA działa podobnie — przemieszcza się po przestrzeni parametrów, testując różne wartości i stopniowo poprawiając wynik, aż znajdzie najlepszy.
Jesteś teraz gotowy/gotowa do przeprowadzenia obliczeń VQE. W tym celu spróbuj rozwiązać poniższe pytanie sprawdzające, które podsumowuje cały proces.
Sprawdź swoją wiedzę
Uzupełnij luki właściwymi terminami, aby dokończyć opis procesu VQE.
VQE to wariacyjny algorytm kwantowy, który łączy moc (1) ________ z obliczeniami klasycznymi i służy do wyznaczania (2) __________ cząsteczki. Proces zaczyna się od zdefiniowania (3) __________, który reprezentuje całkowitą energię układu i pełni rolę obserwowalnego w pomiarach kwantowych. Następnie przygotowujemy (4) __________ — Circuit kwantowy z regulowanymi parametrami, reprezentujący próbną funkcję falową cząsteczki. Parametry te są optymalizowane przy użyciu (5) __________ — klasycznego algorytmu, który iteracyjnie dostosowuje parametry w celu minimalizacji zmierzonej energii. W powyższym omówieniu użyliśmy optymalizatora (6) __________, który doprecyzowuje parametry ansatzu bez potrzeby obliczania pochodnych. Proces trwa aż do osiągnięcia (7) __________, co oznacza, że znaleźliśmy jak najniższą energię cząsteczki.
Bank słów:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Odpowiedź:
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
Obliczanie energii stanu podstawowego atomu wodoru za pomocą VQE
Teraz wykorzystajmy to, czego się nauczyliśmy, do obliczenia energii stanu podstawowego atomu wodoru. W całym module będziemy korzystać z frameworku do obliczeń kwantowych zwanego „wzorcami Qiskit" (ang. Qiskit patterns), który dzieli przepływ pracy na następujące kroki:
- Krok 1: Odwzorowanie klasycznych danych wejściowych na problem kwantowy
- Krok 2: Optymalizacja problemu pod kątem wykonania kwantowego
- Krok 3: Wykonanie przy użyciu prymitywów Qiskit Runtime
- Krok 4: Postprocesowanie i analiza klasyczna

Generalnie będziemy postępować zgodnie z tymi krokami.
Zacznijmy od załadowania niezbędnych pakietów, w tym prymitywów Qiskit Runtime. Wybierzemy też najmniej obciążony dostępny komputer kwantowy.
Poniżej znajduje się kod służący do jednorazowego zapisania swoich danych uwierzytelniających. Pamiętaj, aby usunąć te informacje z notatnika po zapisaniu ich w swoim środowisku, żeby przypadkowo ich nie udostępnić podczas dzielenia się notatnikiem. Więcej wskazówek znajdziesz w przewodnikach Set up your IBM Cloud account oraz Initialize the service in an untrusted environment.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
Poniższa komórka pozwoli ci przełączać się między symulatorem a prawdziwym sprzętem w trakcie pracy z notatnikiem. Zalecamy uruchomienie jej teraz:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Krok 1: Odwzorowanie problemu na obwody kwantowe i operatory
Obliczenia VQE zaczynamy od zdefiniowania hamiltonianu dla cząsteczki wodoru () przy określonej odległości wiązania. Ten hamiltoniam reprezentuje całkowitą energię układu w postaci operatorów kubitowych; został wyprowadzony i odwzorowany z układu molekularnego przy użyciu standardowej procedury: 1) zastosowanie zestawu bazowego STO-6G (konkretnego zbioru funkcji matematycznych służących do przybliżenia orbitali elektronowych), 2) zastosowanie odwzorowania Jordana–Wignera (techniki translacji fermiońskich operatorów opisujących elektrony na operatory kubitowe), 3) redukcja kubitów z wykorzystaniem symetrii hamiltonianu w celu uproszczenia problemu.
Jak wyjaśniliśmy wcześniej, obliczone energie stanu podstawowego są silnie uzależnione od wyboru zestawu bazowego i geometrii cząsteczki (np. odległości wiązania). Dla tej konkretnej konfiguracji i po wykonaniu powyższych transformacji wynikowy hamiltoniam kubitowy jest prosty:
Tutaj reprezentuje operator jednostkowy, a — operator Pauliego-Z działający na jednym kubicie. Współczynniki wynikają z całek obliczonych przy użyciu zestawu bazowego STO-6G dla tej konkretnej odległości wiązania z odpowiednią transformacją.
Mając zdefiniowany hamiltoniam, możemy teraz użyć VQE do obliczenia jego energii stanu podstawowego. Warto porównać obliczoną energię stanu podstawowego z oczekiwanymi wartościami. Dla pojedynczego, izolowanego atomu wodoru (H) energia stanu podstawowego wynosi dokładnie -0,5 hartree (przy braku efektów relatywistycznych). Obliczmy dokładną energię stanu podstawowego naszego konkretnego hamiltonianu kubitowego zdefiniowanego powyżej i porównajmy ją z odpowiednimi znanymi wartościami.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Następnie potrzebujemy sparametryzowanego obwodu kwantowego — ansatz — aby przygotować próbną funkcję falową dla stanu podstawowego. Celem jest znalezienie parametrów , które minimalizują wartość oczekiwaną energii . Wybór ansatz jest kluczowy, ponieważ determinuje zbiór możliwych stanów kwantowych, jakie może przygotować nasz obwód. „Dobry" ansatz jest na tyle elastyczny, żeby móc reprezentować stan bardzo bliski prawdziwemu stanowi podstawowemu badanego hamiltonianu, ale nie na tyle złożony, by wymagał zbyt wielu parametrów lub zbyt głębokiego Circuit dla obecnych komputerów kwantowych.
Tutaj wypróbujemy trzy różne jednokulbitowe ansätze, aby sprawdzić, który z nich zapewnia lepsze „pokrycie" możliwych stanów kwantowych, w jakich może znajdować się pojedynczy qubit. „Pokrycie" oznacza zakres stanów kwantowych, jakie Circuit ansatz może wytwarzać przez zmianę swoich parametrów.
Użyjemy trzech ansätze opartych na różnych kombinacjach jednokulbitowych bramek obrotowych:
- Ansatz z jedną bramką obrotową wokół 1 osi: Ten ansatz używa obrotów tylko wokół jednej osi (). Na sferze Blocha odpowiada to poruszaniu się wyłącznie wzdłuż jednego okręgu. Jest to najmniej elastyczny ansatz, obejmujący ograniczony zbiór stanów.
- Dwa ansätze z bramkami obrotowymi wokół 2 osi: Te ansätze łączą obroty wokół dwóch różnych osi ( oraz ). Pozwala to dotrzeć do większej części sfery Blocha w porównaniu z obrotem wokół jednej osi.
Porównując wyniki VQE uzyskane za pomocą tych trzech ansätze, możemy zobaczyć, jak elastyczność i pokrycie przestrzeni stanów przez ansatz wpływają na naszą zdolność do znalezienia prawdziwej energii stanu podstawowego naszego uproszczonego hamiltonianu. Bardziej elastyczny ansatz ma potencjał znalezienia lepszego przybliżenia, ale może też być trudniejszy dla klasycznego optymalizatora.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Teraz wygenerujmy 5000 losowych liczb dla każdego parametru i wyrysujmy rozkład losowych stanów kwantowych wytworzonych przez trzy ansätze z tymi losowymi parametrami. Możesz myśleć o tych parametrach jak o obrotach wokół różnych osi na powierzchni sfery. Aby zobaczyć rozkład stanów kwantowych, posłużymy się sferą Blocha — trójwymiarową sferą przedstawiającą stan pojedynczego qubit. Każdy punkt na sferze reprezentuje możliwy stan kubitu, przy czym biegunie północny i południowy odpowiadają klasycznym „0" i „1", lecz qubit może też znajdować się gdziekolwiek pomiędzy nimi, wykazując szczególne własności kwantowe, takie jak superpozycja. Najpierw przygotuj niezbędne funkcje do wyrysowania trójwymiarowej sfery Blocha i wygeneruj 5000 losowych parametrów.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Sprawdźmy, jak działa nasz pierwszy ansatz.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
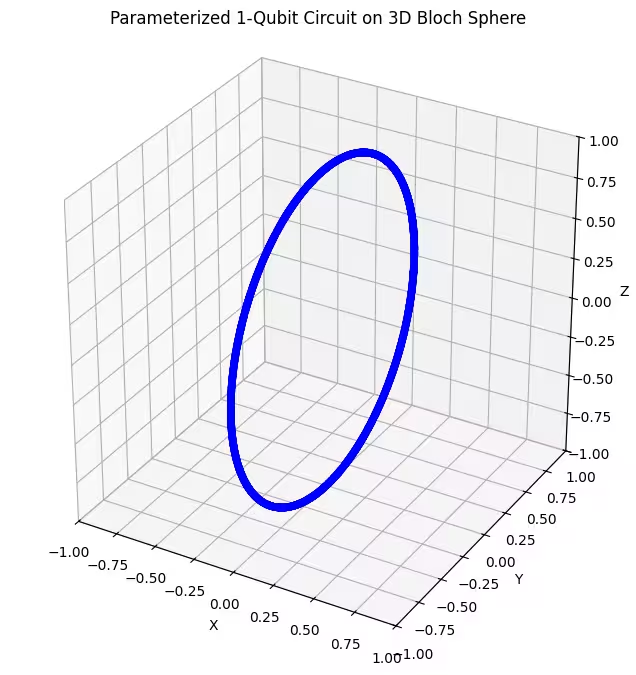
Widzimy, że nasz pierwszy ansatz zwraca stany kwantowe rozłożone w kształcie pierścienia na sferze Blocha. Ma to sens, ponieważ podaliśmy ansatz tylko jeden parametr obrotowy. Może on zatem generować jedynie stany obrócone wokół jednej osi. Startując z punktu i obracając się wokół jednej osi, zawsze otrzymamy pierścień. Sprawdźmy teraz nasz drugi ansatz, który ma dwie ortogonalne bramki obrotowe — Rx i Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
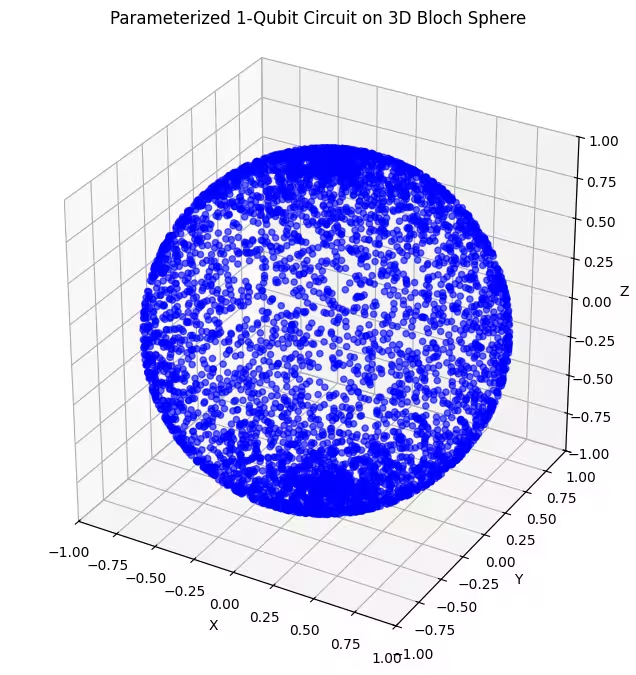
Widzimy, że nasz drugi ansatz pokrywa większy obszar sfery Blocha — zwróć jednak uwagę, że punkty są bardziej zagęszczone wokół biegunów i bardziej rozłożone wokół równika. Czas sprawdzić nasz ostatni ansatz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
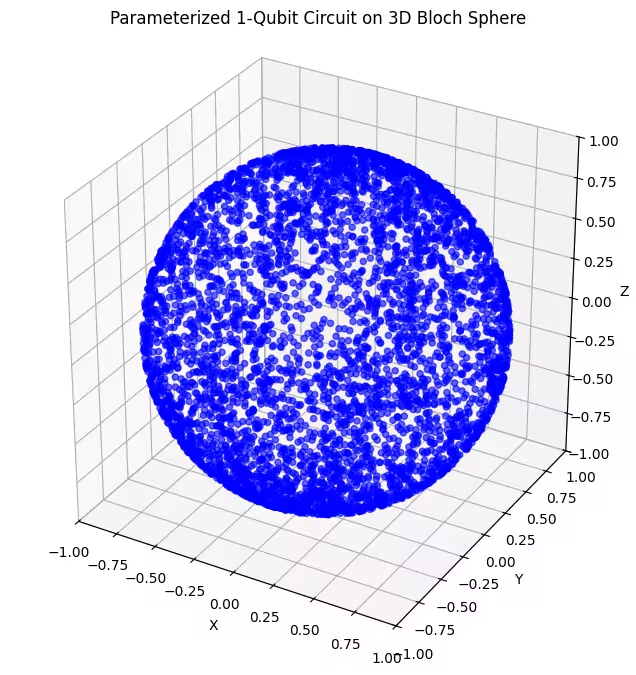
Tutaj widać bardziej równomiernie rozłożone stany kwantowe generowane przez nasz ostatni ansatz.
Jak wspomniałem, najlepszym podejściem jest zdobycie wiedzy o szukanym stanie podstawowym i wybranie ansatz, który dobrze nadaje się do eksploracji stanów bliskich temu stanowi podstawowemu. Na przykład, gdybyśmy wiedzieli, że nasz stan podstawowy leży blisko bieguna, moglibyśmy wybrać ansatz 2. Dla uproszczenia zostaniemy przy ansatz 3, który równomiernie próbkuje całą sferę Blocha.
Teraz, gdy wybraliśmy nasz ansatz, narysujmy Circuit.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Krok 2: Optymalizacja pod kątem docelowego sprzętu
Uruchamiając obliczenia na prawdziwym komputerze kwantowym, nie zależy nam tylko na logice obwodu kwantowego. Ważne są też takie kwestie jak to, jakie operacje może wykonywać dany komputer kwantowy, oraz gdzie na nim znajdują się qubit-y, których używamy. Czy sąsiadują ze sobą? Czy są od siebie odległe? Dlatego następnym krokiem jest przepisanie naszego obwodu przy użyciu bramek naturalnych dla danego komputera kwantowego, z uwzględnieniem rozmieszczenia qubit-ów. Można to osiągnąć przez transpilację — po tym procesie zobaczysz, jak nasz prosty ansatz zostaje przekształcony w inny zestaw bramek, a nasze abstrakcyjne qubit-y zostaną odwzorowane na fizyczne qubit-y w prawdziwym komputerze kwantowym.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Widać, że bramki rx, rz naszego ansatzu zostały przekształcone w ciąg bramek rz, sx, które są natywnymi bramkami naszego Backend-u. Ponadto widać, że nasz q0 jest teraz odwzorowany na piąty fizyczny qubit. Musimy też zaktualizować nasz Hamiltonian zgodnie z tymi zmianami, tak jak w poniższym kodzie:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Krok 3: Wykonanie na docelowym sprzęcie
Nadszedł czas, aby uruchomić nasz VQE na prawdziwym QPU. W tym celu potrzebujemy najpierw funkcji kosztu dla procesu optymalizacji, która wyznacza wartość oczekiwaną Hamiltonianu dla stanu kwantowego generowanego przez ansatz. Nie martw się! Nie musisz kodować wszystkiego samodzielnie. Przygotowaliśmy w tym celu funkcję — wystarczy, że uruchomisz poniższą komórkę.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Na koniec przygotowujemy parametry początkowe dla naszego ansatzu i jego procesu optymalizacji. Możesz po prostu użyć samych zer lub wartości losowych. Poniżej wybraliśmy parametry początkowe, ale możesz dowolnie komentować lub odkomentowywać linie w komórce, aby losować parametry jednostajnie z zakresu od 0 do .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Gratulacje! Właśnie z powodzeniem ukończyłeś swój pierwszy eksperyment z chemii kwantowej. Widzimy różnicę między dokładną energią stanu podstawowego Hamiltonianu a naszym wynikiem, jednak dzięki zastosowaniu domyślnej techniki mitygacji błędów (która koryguje błędy odczytu) różnica jest niewielka. To bardzo dobry początek!
Uwaga: Lepszy wynik możesz uzyskać, ustawiając poziom mitygacji błędów za pomocą resilience_level. Domyślna wartość to 1 — jeśli ustawisz wyższą wartość, zużyje to więcej czasu QPU, ale może zwrócić lepszy wynik.
Krok 4: Post-processing
Czas przyjrzeć się temu, jak działał nasz klasyczny optymalizator. Uruchom poniższą komórkę i sprawdź wzorzec zbieżności.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Zaczęliśmy od dość dobrej wartości początkowej, dzięki czemu uzyskaliśmy dobrą wartość końcową już w zaledwie 10 krokach. Widać duże i małe szczyty — to typowa cecha optymalizatora COBYLA: przeszukuje przestrzeń jak gdyby nie widząc jej krajobrazu i dostosowuje rozmiar kroku przy każdym pomiarze.
Sprawdź swoją wiedzę
Jakie są twoje obserwacje? Który element powyższego procesu można ulepszyć, aby uzyskać wyniki bliższe wartościom teoretycznym lub dokładnej energii stanu podstawowego Hamiltonianu? Co należy wziąć pod uwagę?
Odpowiedź:
Pierwszą rzeczą do rozważenia jest zmiana zestawu baz używanych do obliczania Hamiltonianu cząsteczek. Jak wspomniano wcześniej, energia stanu podstawowego atomu H wynosi -0,5 Hartree — jest to wartość dobrze znana — a wybrana przez nas baza STO-6G nie jest wystarczająca do dokładnego wyznaczenia tej wartości.
Wybór bardziej złożonego rodzaju bazy zwiększa liczbę qubit-ów używanych przez Hamiltonian; dlatego musimy wybrać bardziej złożony i odpowiedni ansatz dla problemów chemicznych.
Kolejnym elementem do optymalizacji jest zarządzanie szumem w QPU. Bardziej zaawansowane techniki mitygacji błędów dają lepsze wyniki, ale mogą wymagać więcej czasu. Warto też rozważyć, jak shot_number wpływa na wyniki.
Na koniec: lepszą zbieżność można też osiągnąć, wypróbowując różne optymalizatory.
Oblicz energię stanu podstawowego cząsteczki wodoru za pomocą VQE
Teraz, gdy zapoznaliśmy się z ogólnym procesem VQE na przykładzie atomów , obliczymy energię stanu podstawowego cząsteczki szybciej.
Krok 1: Odwzorowanie problemu na obwody kwantowe i operatory
Dostarczamy tutaj jednocząstkowy Hamiltonian korzystający z bazy STO-6G oraz transformacji Jordana-Wignera, z redukcją qubitów przez zastosowanie symetrii Hamiltonianu. Zwróć uwagę, że odległość atomowa między dwoma atomami wodoru wynosi 0.735 .
W odróżnieniu od obliczania energii stanu podstawowego pojedynczego atomu wodoru (), wyznaczenie stanu podstawowego cząsteczki wodoru () wymaga uwzględnienia – oprócz energii związanej z orbitalami elektronowymi – siły odpychania działającej między jądrami obu atomów wodoru. W tym kroku podajemy tę wartość jako stałą; jej faktyczne obliczenie będzie zadaniem sprawdzającym.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Krok 2: Optymalizacja pod kątem docelowego sprzętu
Ponieważ liczba qubitów używanych przez poprzedni VQE i Hamiltonian jest taka sama jak w przypadku Backend'u przeznaczonego do wykonania, skorzystamy z istniejącego ansatzu i jego zoptymalizowanej postaci.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Krok 3: Wykonanie na docelowym sprzęcie
Czas na obliczenia na prawdziwym QPU. Niemal wszystko pozostaje bez zmian, ale dobierzemy odpowiedni punkt startowy, aby dopasować go do Hamiltonianu. Ponadto w części iteracyjnej niektóre ustawienia Estimatora, służącego do wyznaczania wartości oczekiwanych Hamiltonianu dla ansatzu na QPU, będą nieco inne niż w poprzednich obliczeniach. Tę zmianę omówimy szerzej w pytaniu sprawdzającym.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Choć VQE teoretycznie zapewnia górne ograniczenie prawdziwej energii stanu podstawowego, praktyczne implementacje na rzeczywistym lub symulowanym z szumem sprzęcie kwantowym, a także przybliżenia przyjęte podczas przygotowywania Hamiltonianu (takie jak zestawy baz lub redukcja qubitów), mogą wprowadzać błędy, które niekiedy skutkują zmierzoną energią nieco niższą od dokładnej wartości teoretycznej lub konkretnej referencji numerycznej. Mimo pewnych błędów wyniki wydają się zadowalające, zwłaszcza biorąc pod uwagę małą liczbę kroków. Zakończmy teraz to obliczenie VQE, przyglądając się temu, jak działał optymalizator.
Krok 4: Post-procesowanie
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Sprawdź swoje zrozumienie
Obliczmy energię odpychania jądrowego cząsteczki , którą uwzględniliśmy jako wartość stałą (0,71997 Hartree).

Skorzystaj z prawa Coulomba i jednostek atomowych, aby upewnić się, że wynik jest w Hartree.
Hartree.Odpowiedź:
Ponieważ oba jądra wodoru są naładowane dodatnio, odpychają się wzajemnie siłą elektrostatyczną. To odpychanie opisuje prawo Coulomba:
,
gdzie to ładunek protonu, to przenikalność elektryczna próżni, a to odległość między dwoma jądrami, mierzona w metrach lub promieniach Bohra w jednostkach dżuli (J).
Aby obliczyć tę energię w Hartree, należy przepisać powyższe równanie w układzie jednostek atomowych (AU). W AU , , a promień Bohra () wynosi 1 i staje się podstawową skalą długości w AU. Dzięki tym uproszczeniom prawo Coulomba redukuje się do:
,
gdzie musi być mierzony w promieniach Bohra ().
Aby przeliczyć podaną odległość jądrową z na , potrzebujemy następującej relacji przeliczeniowej:
zatem odpowiada .
W związku z tym energia odpychania jądrowego dla danego wynosi
Obliczanie energii reakcji
Teraz użyjmy tego, co uzyskaliśmy! Wykorzystałeś VQE — wariacjonalny solver kwantowy — do obliczenia energii stanu podstawowego atomu oraz cząsteczki . Pozostało jeszcze wykorzystanie obliczonych wartości do wyznaczenia energii reakcji procesu .
Energia reakcji to zmiana energii, która zachodzi, gdy substancje reagują ze sobą, tworząc nowe substancje. Wyobraź sobie, że coś budujesz: niekiedy musisz włożyć w to energię (jak układanie klocków), a niekiedy energia jest uwalniana (jak piłka tocząca się w dół zbocza). W chemii reakcje albo pochłaniają energię (reakcje endotermiczne), albo ją uwalniają (reakcje egzotermiczne).
Energię reakcji procesu można obliczyć według następującego wzoru:
Uruchamiając poniższą komórkę, zobaczmy to wizualnie. Użyjemy dokładnej wartości energii stanu podstawowego każdego Hamiltonianu i porównamy energię reakcji dla dokładnego rozwiązania oraz wyników VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Jak widać na rysunku, mimo pewnych błędów, dokładna energia stanu podstawowego Hamiltonianów oraz energia reakcji obliczona przy użyciu wyników VQE są zbliżone — wynoszą około -0,2 Hartree.
Należy tu zaznaczyć, że energia reakcji tego procesu ma wartość ujemną, co oznacza, że energia jest uwalniana w toku reakcji, a powstała cząsteczka ma niższą energię niż dwa pojedyncze atomy. 6. Wnioski
Podsumujmy to, czego się do tej pory nauczyliśmy.
Na początku przyjrzeliśmy się dwóm ważnym technikom aproksymacji niezbędnym do rozwiązywania zagadnień chemii kwantowej: zasadzie wariacyjnej oraz wyborom zbiorów bazowych — obu fundamentalnych dla VQE. Zasadę wariacyjną zbadaliśmy ręcznie, obliczając energię stanu podstawowego prostego oscylatora harmonicznego.
Następnie omówiliśmy VQE — powszechnie stosowany algorytm do obliczania energii stanu podstawowego układu kwantowego. Uruchomiliśmy kod obliczający energie stanu podstawowego atomowego wodoru () oraz cząsteczki wodoru (). W szczególności dowiedzieliśmy się, że konieczne jest uzyskanie odpowiedniego molekularnego Hamiltonianu dla danego układu i przekształcenie go do postaci wykonywalnej na komputerze kwantowym. Zobaczyliśmy też, że ansatz — sparametryzowany Circuit kwantowy — jest potrzebny do przygotowania próbnych stanów kwantowych w VQE, oraz omówiliśmy znaczenie wyboru odpowiedniej struktury Circuit ansatzu. Dowiedzieliśmy się również, że VQE opiera się na iteracyjnym procesie optymalizacji z użyciem klasycznego komputera, który kieruje Circuit kwantowym w poszukiwaniu stanu o najniższej energii, i zobaczyliśmy, jak ten proces zbiega się.
Na koniec użyliśmy obliczonych przez VQE energii stanu podstawowego i do wyznaczenia energii reakcji procesu .
VQE jest potężnym algorytmem kwantowym bliskiej perspektywy, lecz ważne jest, by zdawać sobie sprawę z jego ograniczeń. Wydajność VQE w dużej mierze zależy od wyboru ansatzu — znalezienie efektywnie przygotowywalnego ansatzu, który dokładnie reprezentuje prawdziwy stan podstawowy, staje się coraz trudniejsze dla większych, bardziej złożonych cząsteczek. Ponadto obecny sprzęt kwantowy jest podatny na szumy, co może wpływać na dokładność wyników VQE, szczególnie dla głębszych Circuitów lub większej liczby Qubitów. Mimo tych wyzwań VQE stanowi fundamentalny algorytm, a trwające badania eksplorują bardziej zaawansowane metody wariacyjne oraz techniki mitygacji błędów, aby poszerzać granice możliwości w chemii kwantowej na komputerach kwantowych bliskiej perspektywy. Na przykład opracowywane są algorytmy takie jak Sample-based Quantum Diagonalization (SQD), które wykorzystują próbki uzyskane z Circuitów kwantowych w połączeniu z klasyczną diagonalizacją w podprzestrzeni, aby poprawić estymację energii i rozwiązać niektóre ograniczenia VQE — w szczególności dotyczące wydajności pomiarów i odporności na szumy.
Powtórzenie i pytania
Kluczowe pojęcia:
- Wariacyjny algorytm kwantowy to paradygmat obliczeniowy, w którym klasyczny komputer i komputer kwantowy współpracują ze sobą przy rozwiązywaniu problemu.
- W VQE zaczynamy od Hamiltonianu układu i odwzorowujemy go na Qubitach w celu wykonania na komputerze kwantowym. Wybieramy sparametryzowany Circuit kwantowy — ansatz — i wielokrotnie wykonujemy pomiary, zmieniając parametry ansatzu, aż do osiągnięcia najniższej wartości energii. Przeszukiwanie przestrzeni parametrów odbywa się przy użyciu klasycznego optymalizatora. Aby uzyskać dobre wyniki, konieczny jest dobór odpowiedniego ansatzu i właściwego optymalizatora.
- Energia reakcji to całkowita zmiana energii w reakcji chemicznej, wyznaczana przez różnicę między energią substratów a energią produktów.
Prawda/fałsz
- Zasada wariacyjna stwierdza, że wartość oczekiwana energii dla dowolnej próbnej funkcji falowej jest zawsze większa lub równa prawdziwej energii stanu podstawowego.
- Zbiór bazowy to zbiór funkcji służących do aproksymacji kwantowych funkcji falowych.
- VQE jest algorytmem kwantowym służącym do dokładnego rozwiązywania równania Schrödingera dla danego Hamiltonianu.
- W VQE sparametryzowany Circuit kwantowy (ansatz) jest używany do przygotowania próbnych funkcji falowych.
- Wybór optymalizatora w VQE (na przykład COBYLA, SPSA lub ADAM) nie wpływa na jakość wyników.
- Qiskit's
Estimatorjest używany do bezpośredniego obliczania wartości oczekiwanych Hamiltonianów w VQE.
Pytania wielokrotnego wyboru:
- Jaka jest rola Hamiltonianu w VQE?
- A) Generowanie losowych stanów kwantowych
- B) Wyznaczanie energii stanów kwantowych
- C) Optymalizowanie Circuitów kwantowych
- D) Tworzenie splątania
- Jaki jest główny cel algorytmu VQE?
- A) Znalezienie energii stanu podstawowego Hamiltonianu
- B) Tworzenie splątania między Qubitami
- C) Wykonywanie wyszukiwania Grovera
- D) Łamanie szyfrowania RSA
- Ile stanów kwantowych jest generowanych w tym notebooku w celu porównania ansatzu?
- A) 100
- B) 1000
- C) 5000
- D) 10 000
- Dlaczego w VQE wymagany jest klasyczny optymalizator?
- A) Do wykonywania pomiarów kwantowych
- B) Do aktualizowania parametrów ansatzu w celu minimalizacji energii
- C) Do splątywania Qubitów
- D) Do generowania kwantowej losowości
- Dlaczego ansatz jest zaprojektowany jako sparametryzowany?
- A) Aby umożliwić przygotowanie stanów kwantowych
- B) Aby umożliwić przeszukiwanie szerokiej przestrzeni stanów kwantowych
- C) Aby zmniejszyć złożoność Circuit
- D) Aby bezpośrednio mierzyć wartości własne
- Które z poniższych stwierdzeń jest najbardziej trafne w odniesieniu do wyboru dobrego ansatzu?
- A) Ansatz musi produkować stany równomiernie rozłożone na sferze Blocha, w przeciwnym razie zawiedzie.
- B) Ansatz powinien być dopasowany do danego układu, aby mieć pewność, że może generować stany bliskie stanowi podstawowemu.
- C) Ansatz powinien produkować losowe stany przy użyciu swoich parametrów wariacyjnych.
- D) Lepszy ansatz zawsze ma więcej parametrów wariacyjnych.
(Opcjonalnie) Dodatek: Narzut optymalizatora a złożoność ansatzu
VQE napotyka kilka dobrze znanych wyzwań[ref 6], a poniższe są związane z tym, czego się już nauczyliśmy.
- Wyzwania związane z doborem ansatzu
Wybór odpowiedniego ansatzu wariacyjnego stanowi inherentny problem. Ansätze inspirowane chemią (jak UCCSD) zapewniają fizyczną dokładność, lecz wymagają głębokich układów, podczas gdy ansätze wydajne sprzętowo mają płytsze układy, ale mogą brakować im fizycznej interpretowalności. Ponadto wiele ansätzów wprowadza nadmiarowe parametry wariacyjne, które w małym stopniu przyczyniają się do poprawy dokładności, za to znacząco utrudniają optymalizację.
- Trudności optymalizacyjne
Przestrzeń optymalizacji VQE może zawierać obszary, w których gradienty zanikają wykładniczo (tzw. „barren plateaus"), co utrudnia klasycznym optymalizatorom efektywną aktualizację parametrów wariacyjnych. W odpowiedzi na to badacze próbowali stosować różne rodzaje optymalizatorów – gradientowe i bezgradientowe – jednak oba podejścia napotykają trudności. Optymalizatory gradientowe cierpią na problem barren plateaus, natomiast metody bezgradientowe wymagają dużej liczby wywołań funkcji.
- Narzut optymalizatora
Kolejnym dobrze znany wyzwaniem jest narzut optymalizatora, związany ze skalą problemu. Układy kwantowe wymagane przez VQE rosną w głębokości i złożoności wraz ze wzrostem rozmiaru problemu; typowo zwiększa to też liczbę parametrów do optymalizacji. Proces optymalizacji staje się niewykonalny w miarę wzrostu liczby parametrów, co prowadzi do wolnej zbieżności i trudności w znalezieniu optymalnego rozwiązania.
Poniżej przyjrzymy się tym wyzwaniom, stosując VQE dla cząsteczki z dwoma różnymi rodzajami ansätzów.
(Uwaga: Może to zająć więcej czasu QPU, więc jeśli nie masz wystarczająco czasu, możesz skorzystać z symulatora.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Teraz uruchomimy VQE z punktem startowym złożonym z samych jedynek, z maksymalnie 20 krokami, i porównamy zbieżność obu przebiegów.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Powyższy wykres wyraźnie pokazuje, że proces optymalizacji ansatzu z większą liczbą zmiennych potrzebuje więcej czasu, aby osiągnąć stabilną zbieżność.
Zamiast polegać na prostych układach jednoQubitowych i nieskomplikowanym ansatzu, złożoność optymalizacji rośnie, gdy wymagane są większe układy kwantowe i bardziej złożone struktury ansätzów. Uwypukla to dobrze znane wyzwanie w VQE: narzut optymalizatora.
Badacze wciąż opracowują różne zaawansowane metodologie, które mogą wykorzystywać komputery kwantowe do rozwiązywania problemów chemicznych. Możesz uzyskać dostęp do różnorodnych materiałów edukacyjnych na stronie IBM Quantum Learning.
Referencje
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)